HOME м•ҢлҰјмҶҢмӢқ кё°мҲ лҸҷн–Ҙ
| [мғқл¬јн•ҷм—°кө¬м •ліҙм„јн„°(BRIC)] нҳ„мғҒн•ҷл¶Җн„° л©”м»ӨлӢҲмҰҳк№Ңм§Җ лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷмқҳ нҷ•мһҘ |
| мқҙлҰ„ : н‘ңмӨҖм„ұкіјн•ҷмӮ°нҢҖ | мһ‘м„ұмқј : 2018.01.25 | мЎ°нҡҢмҲҳ : 22804 |
| кё°кҙҖ : мғқл¬јн•ҷм—°кө¬м •ліҙм„јн„°(BRIC) |
|
нҳ„мғҒн•ҷл¶Җн„° л©”м»ӨлӢҲмҰҳк№Ңм§Җ лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷмқҳ нҷ•мһҘ
[мҡ”м•Ҫл¬ё]
мғқл¬јн•ҷм—җм„ң к°ҖмһҘ кё°ліём Ғмқё м„ё к°Җм§Җ к¶ҒкёҲмҰқмқҖ мІ«м§ё, м–ҙл–»кІҢ к°ңлі„ м„ёнҸ¬л“Өмқҙ 분нҷ”н•ҳм—¬ мЎ°м§Ғмқ„ нҳ•м„ұн•ҳлҠ”м§Җ, л‘ҳм§ё, м–ҙл– н•ң л°©мӢқмңјлЎң мЎ°м§Ғмқҙ кё°лҠҘмқ„ мҲҳн–үн•ҳлҠ”м§Җ, м…Ӣм§ё, м–ҙл–Ө мң м „мһҗ мЎ°м Ҳ л©”м»ӨлӢҲмҰҳмқҙ мқҙлҹ¬н•ң кіјм •л“Өмқ„ л’·л°ӣм№Ён•ҳлҠ”м§Җ мқҙлӢӨ. мқҙлҹ¬н•ң к¶ҒкёҲ м җл“Өмқ„ н•ҙкІ°н•ҳкё° мң„н•ҙ, лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷ(single-cell genomics)мқҙ мғҲлЎңмҡҙ кёёмқ„ м—ҙм–ҙмЈјкі мһҲлӢӨ. мқҙлҠ” мң м „мІҙн•ҷмқҳ мў…н•©м Ғмқё нҠ№м„ұкіј нҳ„лҜёкІҪм Ғ н•ҙмғҒлҸ„(resolution)лҘј кІ°н•©н•ЁмңјлЎңмҚЁ, ліөмһЎн•ң лӢӨм„ёнҸ¬ мӢңмҠӨн…ңмқ„ м„ӨлӘ…н•ҳкё° мң„н•ҙ н•„мҲҳм ҒмқҙлӢӨ. мҙҲкё°мқҳ лӢЁмқј м„ёнҸ¬ мң м „мІҙ м—°кө¬л“ӨмқҖ мқҙм§Ҳм Ғмқё м„ёнҸ¬ мғҒнғңм—җ лҢҖн•ң нҳ„мғҒл“Өмқ„ лӢӨмҲҳ м ңмӢңн•ҙмҷ”лӢӨ. к·ёлҹ¬лӮҳ кё°мЎҙмқҳ мЎ°м§Ғ кҙҖм°°м Ғ м—°кө¬м—җм„ң, ліҙлӢӨ м—ӯлҸҷм Ғ, мқёкіјм Ғ л©”м»ӨлӢҲмҰҳ лӘЁлҚё м—°кө¬мқҳ н•„мҡ”м„ұмқҙ м ңкё°лҗҳкі мһҲмңјл©°, мқҙлҘј мң„н•ҙм„ңлҠ” мқҙлЎ м Ғ, кі„мӮ°м Ғ, мӢӨн—ҳм Ғ мІҙм ңмқҳ к°•л Ҙн•ң нҶөн•© м—°кө¬к°Җ мҡ”кө¬лҗңлӢӨ.
[лӘ© м°Ё]
1. м„ңлЎ
лӢӨм„ёнҸ¬ мғқл¬јмІҙл“ӨмқҖ м„ёнҸ¬ к°„мқҳ нҳ‘лҸҷмқ„ мң„н•ң м „лһөл“Өмқ„ л°ңм „мӢңмјңмҷ”лӢӨ. н•ҳлӮҳмқҳ лӢЁмқј мң м „мІҙмқҳ кІҪмҡ°, н•Ёк»ҳ мқјн• л•Ң м Ғн•©м„ұмқ„ к·№лҢҖнҷ”мӢңнӮ¬ мҲҳ мһҲлҸ„лЎқ м „л¬ём Ғмқҙкі мғҒнҳёліҙмҷ„м Ғмқё кё°лҠҘ н”„лЎңк·ёлһЁмқ„ м•”нҳёнҷ”н•ңлӢӨ. м„ёнҸ¬, мЎ°м§Ғ л°Ҹ мһҘкё°мҷҖ к°ҷмқҖ м—¬лҹ¬ лӢЁкі„м—җм„ңмқҳ кө¬нҡҚнҷ”(compartmentalization)лҠ” м„ёнҸ¬мҷҖ мӢңмҠӨн…ңл“Өмқҳ кё°лҠҘм Ғ лӢӨм–‘м„ұмқ„ м•јкё°н•ңлӢӨ. мң м „мІҙмқҳ м„ёнҸ¬м§Ҳ л°Ҹ н•ө кө¬нҡҚм—җм„ң мҶҢ분мһҗ(small-molecule), RNA л°Ҹ лӢЁл°ұм§Ҳ лҶҚлҸ„мқҳ м„ нғқм Ғ кҙҖлҰ¬лҘј нҶөн•ң л°ҳмһҗлҸҷ мқҳмӮ¬ кІ°м • н”„лЎңм„ёмҠӨлҘј мң м§Җн•ҳкё° мң„н•ҙ, л¬јлҰ¬м Ғ ліөм ңмҲҳк°Җ м„ёнҸ¬ лӮҙм—җ лӮҙмһҘлҗҳм–ҙ мһҲлӢӨ. нҠ№м • м„ёнҸ¬ лӮҙмқҳ(intracellular) 분мһҗ кө¬м„ұл¬јмқҳ м җ진м Ғ нҡҚл“қмқ„ нҶөн•ҙ, м„ёнҸ¬мқҳ 분нҷ”к°Җ к°ҖлҠҘн•ҳл©°, м„ёнҸ¬мқҳ кё°м–өмқ„ лӮҳнғҖлӮҙкі мҲҳн–үн•ҳкё° мң„н•ң нӣ„м„ұмң м „мІҙ л©”м»ӨлӢҲмҰҳмқҙ к°ҖлҠҘн•ҳкІҢ н•ңлӢӨ. лҚ” лҶ’мқҖ лӢЁкі„мқҳ мһҘкё°, м„ёнҸ¬л“Ө мӮ¬мқҙмқҳ(intercellular) мӢ нҳё м „лӢ¬, м„ёнҸ¬ л°–мқҳ кө¬мЎ° л°Ҹ нҷҳкІҪм Ғ мӢ нҳёл“ӨмқҖ, м„ёнҸ¬л“Өмқҙ(к·ёлҰ¬кі к·ёл“Өмқҳ мң м „мІҙл“Өмқҙ) л¬јлҰ¬м ҒмңјлЎң лӮҙмһҘлҗң ліөмһЎн•ң кіөк°„м Ғ(spatial) кө¬мЎ°лҘј нҳ•м„ұн•ҳлҠ”лҚ° мӮ¬мҡ©лҗңлӢӨ. лӮҳм•„к°Җ мқҙлҠ” ліөмһЎн•ҳкі кө¬мЎ°нҷ”лҗң мЎ°м§Ғмқ„ м•”нҳёнҷ”н•ҳлҠ” кө¬нҡҚнҷ”(compartmentalization)мқҳ мқҙнӣ„ лӢЁкі„лҘј л§Ңл“ лӢӨ.
лҰ°л„ӨмӢқ мғқл¬ј 분лҘҳлІ•(Linnaean)мқҖ м„ёнҸ¬мқҳ мқҙм§Ҳм„ұм—җ лҢҖн•ң нҳ„мһ¬ мқҙлЎ м Ғ кё°л°ҳмқ„ мқҙлЈЁлҠ” мӨ‘мҡ”н•ң л¶Җ분мқҙлӢӨ. мқҙлҠ” м„ёнҸ¬ мң нҳ•мқҳ 분лҘҳлІ•(taxonomy)мқ„ м •мқҳн•ҳкі , кі„мёөм Ғ 분лҘҳлҘј мғқл¬јмІҙмқҳ л°ңмғқ(ontogeny)м—җ л§һ추м–ҙ м§ҖлҸ„лҘј к·ёлҰ¬кё° мң„н•ң лӘ©м Ғмқ„ к°Җ진лӢӨ. мқҙмҷҖ кҙҖл Ён•ҙ нҳ„лҜёкІҪ л°Ҹ FACS (fluorescence-activated cell sorting)лҘј мӮ¬мҡ©н•ҳм—¬ мЎ°м§Ғ л°Ҹ кө¬нҡҚ лӮҙмқҳ лӢЁмқј м„ёнҸ¬ кө¬м„ұмқ„ нҠ№м§•м§Җм–ҙ진лӢӨ. мқҙлҹ¬н•ң кё°мҲ л“ӨмқҖ лҶ’мқҖ кіөк°„м Ғ(spatial) лҳҗлҠ” м„ёнҸ¬мқҳ н•ҙмғҒлҸ„(resolution)лҘј к°Җм§Җкі мһҲм§Җл§Ң, м „мІҙм Ғ к·ёлҰ¬кі м •лҹүм ҒмңјлЎң м—°кө¬н•ҳкё° м–ҙл Өмҡҙ 분лҘҳ мІҙкі„к°Җ мғқкІЁлӮҳкі мһҲлӢӨ. мқҙм—җ л”°лқј мқҙлҜём§Җ л°Ҹ мң м „мһҗ л°ңнҳ„ лҚ°мқҙн„°мқҳ лҢҖнҳ•м ҖмһҘмҶҢмқҳ 축м Ғмқ„ нҶөн•ҙ л§ҺмқҖ 분мһҗм Ғ 분лҘҳлІ•(molecular taxonomy)л“Өмқҙ кө¬м¶•лҗҳкі мһҲлӢӨ. мқҙлҹ¬н•ң 분лҘҳлІ•л“ӨмқҖ м„ұмқём—җм„ң мөңмҶҢ м„ёнҸ¬ 분нҷ”лҘј н•ҳлҠ” лӘҮлӘҮ мІҙм„ёнҸ¬ мЎ°м§Ғ(мҳҲ, л§қл§ү), к·ёлҰ¬кі мғқл¬јмІҙмқҳ мҲҳлӘ… лӮҙлӮҙ м—ӯлҸҷм Ғмқҙкі м§ҖмҶҚм ҒмңјлЎң мһ¬л¶„нҷ”н•ҳлҠ” мЎ°м§Ғ(мҳҲ, мЎ°нҳҲ мӢңмҠӨн…ң)м—җм„ң м„ұкіөм ҒмңјлЎң мһ…мҰқлҗҳм—ҲлӢӨ.
мқҙмҡ© к°ҖлҠҘн•ң л§Ҳм»Өл“Өмқҳ нҢ”л ҲнҠё, нҳ„лҜёкІҪ л°Ҹ 분мһҗм Ғ мЎ°м§Ғ н”„лЎңнҢҢмқјл§Ғмқҳ мІҳлҰ¬лҹүмқҳ м§ҖмҶҚм Ғмқё м§Ҳм Ғ н–ҘмғҒм—җлҸ„ л¶Ҳкө¬н•ҳкі , лҰ°л„ӨмӢқ мғқл¬ј 분лҘҳлІ•мқҖ м „мІҙ мЎ°м§Ғ л°Ҹ мғқлӘ…мІҙк№Ңм§Җ нҷ•мһҘлҗ л•Ң ліём§Ҳм ҒмңјлЎң л¶Җм Ғм Ҳн• мҲҳ мһҲлӢӨ. нҠ№нһҲ, 분мһҗм Ғ н–үлҸҷ(molecular behavior)мқҳ ліөн•© нҳјн•©л¬ј(complex mixture)мқҙ кіөк°„м Ғ л°Ҹ л¬јлҰ¬м ҒмңјлЎң лӢЁлӢЁнһҲ кІ°н•©лҗ л•Ңл§ҲлӢӨ м ‘к·јлІ•мқҖ лӘЁнҳён•ҙ진лӢӨ. 분мһҗм Ғ л©”м»ӨлӢҲмҰҳл“Өмқҙ м§ҖмҶҚм Ғмқё кІ°кіјл“Өмқ„ л§Ңл“Өм–ҙ лӮј л•Ң, кі„мёөм Ғ 분лҘҳ лҳҗн•ң мһҗм—°мҠӨлҹҪкІҢ м Ғмҡ©н• мҲҳ м—ҶлӢӨ(мҳҲ, л°ңлӢ¬мғҒмқҳ м—°мҶҚмІҙ(developmental continuum), 분нҷ”м—җм„ңмқҳ 분기(branching), лӘҮлӘҮ м„ёнҸ¬ мң нҳ• к°„ н”ҢлқјмҠӨнӢұ м „нҷҳ(plastic transition), нҳ•нғң л°ңмғқм Ғ ліҖнҷ”(morphogen gradients)мқҳ нҷ•мӮ°, м„ёнҸ¬ н”„лЎңк·ёлһЁмқҳ лі‘мқём Ғ, л¶Ҳк·ңм№ҷн•ң м•…нҷ”). мӢӨм ңлЎң, л°ңлӢ¬ мғқл¬јн•ҷ, нҠ№нһҲ л°°м•„ 분нҷ”м—җм„ң, м§ҖмҶҚм Ғмқё ліҖнҷ”м—җ лҢҖн•ң лӘЁлҚёмқҙ мһҳ л°ңлӢ¬лҗҳм–ҙ мһҲлӢӨ. к·ёлҹ¬лӮҳ мқҙлҹ¬н•ң кіјм •мқҙ м–ҙл–»кІҢ к°Ҳлқјм ё м•Ҳм •лҗң мғҒнғңлЎң 분нҷ”лҗҳлҠ”м§ҖлҘј мқҙн•ҙн•ҳлҠ” кІғмқҖ мғҒлӢ№н•ң лҸ„м „кіјм ңмқҙлӢӨ. мӢ¬м§Җм–ҙ м„ёнҸ¬ мң нҳ•мқҳ м •нҷ•н•ң м •мқҳмЎ°м°ЁлҸ„ н•©мқҳм җм—җм„ң л©Җм–ҙм§Җкі мһҲмңјл©°, лӢӨм–‘н•ң 분야м—җм„ң мқҙлҹ° мӨ‘мҡ”н•ң к°ңл…җл“Өмқ„ м„ңлЎң лӢӨлҘҙкІҢ л¬ҳмӮ¬н•ҳкі мһҲлӢӨ.
кі„мҶҚ лҠҳм–ҙлӮҳлҠ” к°ңлі„ м„ёнҸ¬л“Ө мҲҳмқҳ мң м „м Ғ л°Ҹ 분мһҗм Ғ мғҒнғң н”„лЎңнҢҢмқјл§Ғмқҙ к°ҖлҠҘн•ҙм§җм—җ л”°лқј, лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷмқҳ м¶ңнҳ„мқҖ м„ёнҸ¬мқҳ м •мІҙ(identity) л°Ҹ кё°лҠҘмқҳ мғҲлЎңмҡҙ лҚ°мқҙн„° кё°л°ҳ м •мқҳ л°©лІ•мқј мҲҳ мһҲмңјл©°, м„ м—„м Ғ кі„мёө(priori hierarchies) лҳҗлҠ” мқҙм „м—җ м •мқҳлҗң л§Ҳм»Өл“Өм—җ лҚң мқҳмЎҙм ҒмқҙлӢӨ. л”°лқјм„ң лӢЁмқј м„ёнҸ¬м—җм„ң к¶Ғк·№м ҒмңјлЎң л°ңмғқн•ҳлҠ” ліөмһЎн•ң мң м „мІҙ кё°лҠҘл“Өмқ„ лӢӨлЈЁкё° мң„н•ҙ, мң м „мІҙн•ҷмқҳ мҡ©лҸ„к°Җ л№ лҘҙкІҢ ліҖкІҪлҗҳкі мһҲлӢӨ. мқҙлҠ” мӢңк°„м Ғ м—ӯн•ҷ(temporal dynamics), кіөк°„м Ғ кө¬м„ұ(spatial organization) л°Ҹ 분мһҗм Ғ л©”м»ӨлӢҲмҰҳкіј к°ҷмқҖ, мЈјмҡ” 축(axis)мқ„ мһ¬кө¬м„ұн•ҳлҠ” кІғмқ„ лҸ„мҡё мҲҳ мһҲлӢӨ. лӢЁмқј м„ёнҸ¬ мң м „мІҙ 분야мқҳ мЈјлҗң лҸ„м „ кіјм ңлҠ”, м–‘м Ғ л°Ҹ нҸ¬кҙ„м Ғмқё мң м „мІҙмқҳ нһҳмқ„ нҳ„лҜёкІҪм Ғ н•ҙмғҒлҸ„мҷҖ кІ°н•©н•ҳм—¬, м„ёнҸ¬ л°Ҹ мЎ°м§Ғ кё°лҠҘм—җ лҢҖн•ң лӘЁлҚёмқ„ ліҙлӢӨ м •лҹүм Ғмқҙл©° мҳҲмёЎ к°ҖлҠҘн•ң лӘЁлҚёлЎң лҢҖмІҙн•ҳлҠ” кІғмқҙлӢӨ.
2. мөңк·јмқҳ лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷ
мң м „мІҙ м „мһҘмқҳ м „мӮ¬мІҙ н”„лЎңнҢҢмқјл§Ғ л°Ҹ нӣ„м„ұмң м „мІҙн•ҷмқҖ м„ёнҸ¬мқҳ 분мһҗ мғҒнғңлҘј мў…н•©м ҒмңјлЎң мёЎм •н•ҳкё° мң„н•ң кёёмқ„ м—ҙм—ҲлӢӨ. мөңк·јк№Ңм§Җ мң м „мІҙ분м„қмқҖ м„ёнҸ¬л“Өмқҳ мқҙм§Ҳм Ғмқё нҳјн•©л¬јмқ„ н’Җл§Ғ(pooling) н•ҳкұ°лӮҳ 분лҘҳ н•ң нӣ„, л¶Җ분 집лӢЁмқ„ н”„лЎңнҢҢмқјл§Ғн•ҳлҠ” л°©мӢқм—җ мқҳмЎҙн•ҳмҳҖлӢӨ. лІҢнҒ¬(bulk) н”„лЎңнҢҢмқјл§ҒмқҖ нҳјн•©л¬јмқҳ нҸүк· мқ„ м ңкіөн•ҳм—¬, мЎ°м ҲлҗҳлҠ” мң м „мһҗм—җ лҢҖн•ң мң м „мІҙ мҠӨнҒ¬лҰ¬лӢқкіј мӢӨн—ҳ к°„мқҳ кө¬м„ұм Ғ ліҖнҷ”лҠ” к°җм§Җн• мҲҳ мһҲм§Җл§Ң, ліём§Ҳм Ғмқё м„ёнҸ¬мқҳ н”„лЎңк·ёлһЁмқҖ м§Ғм ‘ кө¬лі„н• мҲҳ м—Ҷмңјл©°, 분лҘҳ 집лӢЁ(sorted population) 분м„қмқҖ м•Ңл Ө진 н•ҳмң„ 집лӢЁ л°Ҹ 분лҘҳ нҢЁл„җм—җ н•ңм •м ҒмқҙлӢӨ.
м§ҖлӮң лӘҮ л…„ лҸҷм•Ҳ, лӢЁмқј м„ёнҸ¬ 분м„қмқ„ мң„н•ҙ мң м „мІҙ, м „мӮ¬мІҙ л°Ҹ нӣ„м„ұмң м „мІҙ 분м„қ кІҖм •л“Өмқҙ мһ¬кө¬м„ұлҗҳм—ҲлӢӨ. нҳ„мһ¬ лӢЁмқј м„ёнҸ¬мқҳ RNA, DNA, нһҲмҠӨнҶӨ ліҖнҳ•, м—јмғүм§Ҳ м ‘к·јм„ұ(chromatin accessibility), DNA л©”нӢёнҷ”, н•өл§үн•ҳмёө(nuclear lamina) мғҒнҳёмһ‘мҡ© л°Ҹ м—јмғүмІҙмқҳ мҲҳ축(chromosomal contract)лҝҗл§Ң м•„лӢҲлқј лӢЁл°ұм§Ҳ мӢңк·ёлӢҲміҗ л“ұм—җ лҢҖн•ң мң м „мІҙ м „мһҘмқҳ н”„лЎңнҢҢмқјмқ„ мҲҳм§‘н• мҲҳ мһҲлӢӨ. мҙҲкё° м—°кө¬л“ӨмқҖ мІҳлҰ¬лҹү(throughput; м„ёнҸ¬мқҳ мҲҳ), кІ¬кі м„ұ(robustness; лӢӨм–‘н•ң нҖ„лҰ¬нӢ° мғҳн”Ңм—җм„ңмқҳ м„ұлҠҘ), ліөмһЎм„ұ(com-plexity, к°Ғ м„ёнҸ¬м—җм„ң мәЎмІҳн• мҲҳ мһҲлҠ” лҡңл ·н•ң 분мһҗл“Ө) л°Ҹ м •нҷ•м„ұ(accuracy, л…ёмқҙмҰҲ мҲҳмӨҖ)мқ„ н–ҘмғҒмӢңнӮӨлҠ”лҚ° мӨ‘м җмқ„ л‘җм—Ҳм—ҲлӢӨ.
лӢЁмқј м„ёнҸ¬ RNA мӢңнҖҖмӢұ(RNA-seq)мқҖ мқҙлҹ¬н•ң л°©лІ•л“Өмқҳ м„ л‘җм—җ мһҲлӢӨ(нҠ№нһҲ мІҳлҰ¬лҹү). мҙҲкё° м—°кө¬л“ӨмқҖ мҶҢмҲҳмқҳ м„ёнҸ¬л“Өмқ„ лҢҖмғҒмңјлЎң 분м„қлҗҳм—Ҳм§Җл§Ң, лЎңлҙҮ кіөн•ҷ(robotics), лҜём„ёмң мІҙм—ӯн•ҷ(microfluidics) л°Ҹ м—ӯмң нҷ”м•Ў(reverse emulsion) л°Ҹ н•ҳмқҙл“ңлЎңкІ” л°©мҡё(hydrogel droplets)мқ„ нҸ¬н•Ён•ң мқјл Ёмқҳ кё°мҲ м Ғ 진ліҙлҠ” мӢӨн—ҳлӢ№ мҲҳл§Ң лҳҗлҠ” мҲҳмӢӯл§Ң м„ёнҸ¬л“ӨлЎң 분м„қ мІҳлҰ¬лҹүмқ„ мҰқк°ҖмӢңмј°лӢӨ. м„ёнҸ¬лҘј нҡҚл“қн•ҳкі к·№мҶҢлҹүмқҳ RNAлҘј мІҳлҰ¬н•ҳлҠ” кё°мҲ лҳҗн•ң л°ңм „н–ҲлӢӨ. мқҙлҠ” мғқкІҖ(biopsy)мқҙлӮҳ кі м •лҗң м„ёнҸ¬(fixed cell)мҷҖ к°ҷмқҖ мһ‘мқҖ мғҳн”Ңм—җм„ңмқҳ лӢЁмқј м„ёнҸ¬ RNA-seqмқҳ кІ¬кі м„ұмқ„ н–ҘмғҒмӢңмј°лӢӨ. м•„м§Ғ лӢЁмқј м„ёнҸ¬ RNA-seqмқ„ мһ„мғҒ мғҳн”Ңм—җ м Ғмҡ©н•ҳкё°м—җлҠ” мқҙлҘҙм§Җл§Ң, мқҙнӣ„ 분лӘ…нһҲ к°ҖлҠҘн• кІғмңјлЎң ліҙмқёлӢӨ.
мІҳлҰ¬лҹү, кІ¬кі м„ұкіјлҠ” лӢ¬лҰ¬, лӢЁмқј м„ёнҸ¬ RNA-seqмқҳ ліөмһЎм„ұкіј м •нҷ•м„ұмқҖ мөңм Ғнҷ”н•ҳлҠ”лҚ° м–ҙл ӨмӣҖмқҙ мһҲлӢӨ. лӢЁмқј м„ёнҸ¬ RNA-seqмқҖ мөңмҶҢн•ңмқҳ мҳӨлҘҳлЎң к°Ғ м„ёнҸ¬ лӮҙмқҳ м „л № RNA 분мһҗ(messenger RNA molecule)мқҳ мҷ„м „н•ң мЎ°мӮ¬(census)лҘј лӘ©н‘ңлЎң н•ңлӢӨ. к·ёлҹ¬лӮҳ к°Ғк°Ғмқҳ м„ёнҸ¬лҠ” мқҙм „м—җ м•Ңл Өм§Җм§Җ м•ҠмқҖ к°ҖліҖм Ғмқё mRNA н•Ёлҹү(content)мқ„ к°Җм§Җкі мһҲкі , лӢӨлҘё м„ёнҸ¬м Ғ нҠ№м„ұл“Өмқҙ mRNAмқҳ нҡҢліөм—җ мҳҒн–Ҙмқ„ мӨ„ мҲҳ мһҲкё° л•Ңл¬ём—җ, нҳ„мһ¬мқҳ л°ҳліө мӢӨн—ҳмқ„ нҶөн•ҙм„ңлҠ” 분м„қ(assay) м„ұлҠҘмқ„ нҸүк°Җн•ҳлҠ” кІғмқҖ л¶Ҳк°ҖлҠҘн•ҳлӢӨ. ліөмһЎм„ұ нҸүк°ҖлҠ” м„ёнҸ¬лӢ№ 105-106 분мһҗлЎң мҳҲмёЎлҗң м„ёнҸ¬мқҳ mRNA н•Ёлҹү к°Җм •м—җ кё°л°ҳн•ңлӢӨ. Noise нҸүк°ҖлҠ” лӮ®мқҖ ліҖлҸҷм„ұмқ„ к°–лҠ” м „мӮ¬мІҙмқҳ 분мӮ°(variance) 분м„қ лҳҗлҠ” мҠӨнҢҢмқҙнҒ¬ мқё м»ЁнҠёлЎӨ(spike-in control)лҘј мӮ¬мҡ©н•ҳм—¬ мҲҳн–үлҗңлӢӨ. мҳӨлҘҳ ліҙм •( correction)мқ„ мң„н•ң кі мң н•ң 분мһҗм Ғ мӢқлі„мһҗ(molecular identifier) л°Ҹ кё°мҲ л“ӨмқҖ PCR мӨ‘ліөмқ„ мІҳлҰ¬н•ҳкі , м„ёнҸ¬к°„ мҳӨм—ј(cross-cell contamination)мқ„ нғҗм§Җн•ҳкі , лӢӨмҡҙмҠӨнҠёлҰј нҶөкі„ лӘЁлҚём—җ лҢҖн•ң 분мһҗмҲҳ м •мқҳлҘј н•ЁмңјлЎңмҚЁ кё°мҲ м Ғ л…ёмқҙмҰҲлҘј нҒ¬кІҢ мӨ„мқёлӢӨ. к·ёлҹјм—җлҸ„ л¶Ҳкө¬н•ҳкі лӢЁмқј м„ёнҸ¬ RNA-seqмқҖ н•ң 집лӢЁмқҳ м„ёнҸ¬л“Ө мӨ‘ мқјл¶Җл§Ңмқ„ м§Ғм ‘ мёЎм •н•ҳлҠ” мғҳн”Ңл§Ғ м „лһөмқ„ к·ёлҢҖлЎң н•ңлӢӨ.
мғҳн”Ңл§ҒмқҖ м •нҷ•н•ҳкІҢ м Ғмҡ©лҗ л•Ң нҡЁмңЁм Ғмқё мӢӨн—ҳл””мһҗмқёмқ„ мң„н•ң к°•л Ҙн•ң м ‘к·јл°©мӢқмқҙ лҗ мҲҳ мһҲлӢӨ. м„ёнҸ¬ 집лӢЁ лҳҗлҠ” мЎ°м§Ғм—җ лҢҖн•ң мөңм Ғмқҳ лӢЁмқј м„ёнҸ¬ RNA-seq мғҳн”Ңл§Ғ м „лһөмқҖ м—°кө¬мқҳ лӘ©м Ғм—җ лӢ¬л Ө мһҲлӢӨ. лӢЁмқј м„ёнҸ¬ RNA-seq лқјмқҙлёҢлҹ¬лҰ¬ мӢңнҖҖмӢұмқҳ н•ңкі„ нҡЁмҡ©мқҖ мӢңнҖҖмӢұ к№Ҡмқҙ(depth)м—җ л”°лқј кёүкІ©нһҲ к°җмҶҢн•ңлӢӨ. л•Ңл¬ём—җ м Ғн•©н•ң м„ёнҸ¬ мң нҳ•мқҳ мӢқлі„ л°Ҹ 분лҘҳлҘј мң„н•ҙм„ңлҠ” м„ёнҸ¬лӢ№ лҚ” м ҒмқҖ мҲҳмқҳ лҰ¬л“ң(read)лЎң лҚ” л§ҺмқҖ мҲҳмқҳ м„ёнҸ¬лҘј м—°кө¬н•ҙм•јн•Ёмқҙ лӢӨм–‘н•ң 분м„қкІ°кіјл“ӨлЎңл¶Җн„° м ңмӢңлҗҳкі мһҲлӢӨ. лҢҖлҹүмқҳ м„ёнҸ¬ н”„лЎңнҢҢмқјл§Ғмқ„ нҶөн•ҙ мң мӮ¬н•ң RNA 분нҸ¬лҘј к°–лҠ” м„ёнҸ¬мқҳ нҒҙлҹ¬мҠӨн„°лҘј нҷ•мқён• мҲҳ мһҲмңјл©°, м„ёнҸ¬мқҳ мҲҳм—җ мқҳн•ҙ м ңн•ңлҗҳм—ҲлҚҳ лӢЁмқј м„ёнҸ¬ м „мӮ¬мқҳ мқҙмғҒнҷ”лҗң лӘЁлҚёмқ„ нҳ•м„ұн• мҲҳ мһҲлӢӨ. л°ҳл©ҙм—җ, к°Ғ м„ёнҸ¬лЎңл¶Җн„° мөңмҶҢлҹүмқҳ RNAлҘј м–»м§Җ лӘ»н•ҳл©ҙ мғҳн”Ңл§Ғлҗң м„ёнҸ¬л“Өмқ„ н•ҳмң„ нҒҙлҹ¬мҠӨн„°лЎң к·ёлЈ№нҷ”н•ҳлҠ” кІғмқҖ л¶Ҳк°ҖлҠҘн•ҳлӢӨ. лӢЁмқј м„ёнҸ¬ лӮҙмқҳ мң м „мһҗл“Ө к°„мқҳ мЎ°м Ҳ кҙҖкі„лҘј 분м„қн•ҳкё° мң„н•ҙм„ңлҠ” ліҙлӢӨ нҒ° мғҳн”Ңл§Ғ к№Ҡмқҙк°Җ н•„мҡ”н• кІғмқҙлӢӨ.
мІҳлҰ¬лҹү, кІ¬кі м„ұ л°Ҹ ліөмһЎм„ұмқҖ лӢЁмқј м„ёнҸ¬ нӣ„м„ұмң м „мІҙ 분м„қл“Ө(assay)м—җлҸ„ мөңм Ғнҷ”лҗҳм–ҙ мһҲлӢӨ. нҳ„мһ¬ мқҙлҹ¬н•ң 분м„қ(assay)л“ӨмқҖ мҲҳмӢӯм—җм„ң мҲҳл°ұ к°ңмқҳ м„ёнҸ¬лҘј л¶Җ분 мһҗлҸҷнҷ”лЎң 분м„қн•ңлӢӨ. ліөмһЎм„ұмқҖ нҠ№нһҲ лӢЁмқј м„ёнҸ¬ нӣ„м„ұмң м „мІҙ 분м„қмқҳ лҸ„м „кіјм ңмқҙлӢӨ. лӢЁмқј м„ёнҸ¬ RNA-seqкіјлҠ” лӢ¬лҰ¬, нӣ„м„ұмң м „мІҙ 분м„қмқҖ лӢЁмқј ліөм ңмҲҳ(single-) 분мһҗлҘј нғҖкІҹн•ҙм•ј н•ҳл©°, лҶ’мқҖ ліөм ң мҲҳ (high-) 분мһҗ 분м„қмқ„ нҶөн•ң л¶Җ분м Ғ мғҳн”Ңл§ҒмңјлЎңлҠ” ліҙмҷ„н• мҲҳ м—ҶлӢӨ. м ҖліөмһЎм„ұ лҚ°мқҙн„°лҘј н”јн•ҳкё° мң„н•ҙ л‘җ к°Җм§Җ н’Җл§Ғ м „лһөмқҙ м Ғмҡ©лҗҳм—ҲлӢӨ. мІ« лІҲм§ё м „лһөмқҖ 분мһҗ нҡҢмҲҳмңЁ(molecule recovery rate)мқҙ 1-10%мқј л•Ңм—җлҸ„ нҡЁкіјм Ғмқё 분м„қмқ„ к°ҖлҠҘн•ҳкІҢ н•ҳкё° мң„н•ҙ лӢЁмқј м„ёнҸ¬лҘј лӘЁмңјлҠ” кІғмқҙлӢӨ. л‘җ лІҲм§ё м „лһөмқҖ м—¬лҹ¬ м—°кҙҖлҗң мҳҒм—ӯл“Ө к°„мқҳ к°ҷмқҖ м„ёнҸ¬ лӮҙм—җ мӢ нҳёл“Өмқ„ лӘЁмңјлҠ” кІғмқҙлӢӨ(мҳҲ. к°ҷмқҖ м „мӮ¬ мқёмһҗм—җ мқҳн•ң кІ°н•© мҳҒм—ӯл“Ө). мқҙлҠ”, мҳҒм—ӯлі„ coverageк°Җ лӮ®лҚ”лқјлҸ„ м„ёнҸ¬мқҳ нӣ„м„ұ мғҒнғң(epigenetic state)лҘј нҡҢліө(recovery) н•ҳлҠ”лҚ° нҡЁкіјм ҒмқҙлӢӨ.
DNA л©”нӢёнҷ”, нһҲмҠӨнҶӨ ліҖнҳ•, м—јмғүм§Ҳ м ‘к·јм„ұ л°Ҹ 3D м—јмғүмІҙ кө¬м„ұ(3D chromosome organization)л“ӨмқҖ лӘЁл‘җ кі мң н•ң м •ліҙлҘј к°Җм§Җкі мһҲмңјл©°, мқҙлҠ” мөңлҢҖмқҳ ліөмһЎм„ұкіј мІҳлҰ¬лҹүмқҳ лӢЁмқј м„ёнҸ¬ RNA-seqмңјлЎңлҸ„ м–»мқ„ мҲҳ м—ҶлӢӨ. мҳҲлҘј л“Өм–ҙ, м—јмғүм§Ҳ кө¬м„ұ лӮҙмқҳ ліҖнҷ”лҠ” RNA л°ңнҳ„ лӢЁкі„м—җм„ң м•Ңкё° м „м—җ 분нҷ” нҳ„мғҒмқ„ мҳҲкІ¬н• мҲҳ мһҲмңјл©°, м„ёнҸ¬мқҳ мң нҳ• л°Ҹ м•Ҳм •м„ұм—җ лҢҖн•ң мӢ лў°н• мҲҳ мһҲлҠ” м§Җл¬ё(fingerprint)мқҙ лҗ мҲҳ мһҲлӢӨ. DNA л©”нӢёнҷ” landscapeлҠ” м„ёнҸ¬мқҳ л°ңлӢ¬ к°ҖлҠҘм„ұ(developmental potential)кіј мЎ°м Ҳ мқёмһҗмқҳ нҷңм„ұмқ„ RNA мҲҳмӨҖм—җм„ңлҠ” м¶”лЎ н• мҲҳ м—ҶлҠ” л°©мӢқмңјлЎң л°ҳмҳҒн• мҲҳ мһҲлӢӨ. мөңк·јмқҳ м „лһөл“ӨмқҖ лҸҷмқјн•ң м„ёнҸ¬ лӮҙмқҳ м—¬лҹ¬ мң нҳ•мқҳ н”„лЎңнҢҢмқјмқ„ лҸҷмӢңм—җ мёЎм •н• мҲҳ мһҲлӢӨ. мқҙм—җ м–ҙл–»кІҢ DNAм—җм„ң RNA, лӢЁл°ұм§Ҳ л°Ҹ м„ёнҸ¬мқҳ н‘ңнҳ„нҳ•к№Ңм§Җ л°ңмғқн•ҳлҠ” мЎ°м Ҳ мқҙлІӨнҠёл“Өмқҙ л°ңмғқн•ҳлҠ”м§Җ нҢҢм•…н•ҳлҠ”лҚ° лҸ„мӣҖмқҙ лҗңлӢӨ. л§ҺмқҖ м—°кө¬к°Җ мқҙлҜё DNAмҷҖ RNA, RNAмҷҖ лӢӨм–‘н•ң лӢЁл°ұм§Ҳ мӢңк·ёлӢҲміҗ, RNAмҷҖ DNA л©”нӢёнҷ”мқҳ мёЎм •мқ„ кІ°н•©мӢңмј°мңјл©°, лҚ” лӮҳм•„к°Җ мҢҚ(pairwise) л°Ҹ лӢӨмӨ‘(multiway) мЎ°н•©л“Өмқҳ м—°кө¬к°Җ кі§ мқҙлЈЁм–ҙ м§Ҳ кІғмқҙлӢӨ.
к·ёлҹ¬лҜҖлЎң, лӢЁмқј м„ёнҸ¬ мң м „мІҙ лҚ°мқҙн„°лҠ” к°ҷмқҖ м„ёнҸ¬м—җ лҢҖн•ң мҲҳмІң к°ңмқҳ кі н’Ҳм§Ҳ лӢЁмқј м„ёнҸ¬ RNA-seq н”„лЎңнҢҢмқјкіј лӢЁмқј м„ёнҸ¬ нӣ„м„ұмң м „мІҙ н”„лЎңнҢҢмқјмқ„ нҸ¬кҙ„н• мҲҳ мһҲлӢӨ. мқҙлҹ¬н•ң лҚ°мқҙн„°лҠ” м„ёнҸ¬лҘј нҺён–Ҙлҗҳм§Җ м•Ҡкі нҸ¬кҙ„м ҒмңјлЎң л¶Җ분 집лӢЁмқ„ 분лҘҳн•ҳлҠ” лҸҷмӢңм—җ к°Ғ м„ёнҸ¬ мң нҳ•м—җ лҢҖн•ҙ мң м „мІҙ м „мһҘмқҳ м „мӮ¬ л°Ҹ нӣ„м„ұмң м „мІҙ мғҒнғңлҘј м •мқҳн•ңлӢӨ. к·ёлҹ¬лӮҳ мқҙлҹ¬н•ң лҚ°мқҙн„°мҷҖ к·ё 분лҘҳлҘј мң м „мІҙмҷҖ мЎ°м§Ғ мЎ°м Ҳ(tissue reg-ulation)мқҳ кё°кі„лЎ м Ғ лӘЁлҚёлЎң 분лҘҳн•ҳкё° мң„н•ҙм„ңлҠ”, лӢЁмқј м„ёнҸ¬л“Өмқҙ мӢңк°„м Ғ(temporal), кіөк°„м Ғ(spatial) мғҒнҷ©м—җм„ң мҡ°м„ мёЎм •лҗҳкі н•ҙм„қлҗҳм–ҙм•ј н•ңлӢӨ.
3. мӢңк°„м Ғ 축 л°Ҹ м—ӯн•ҷмқҳ 추лЎ
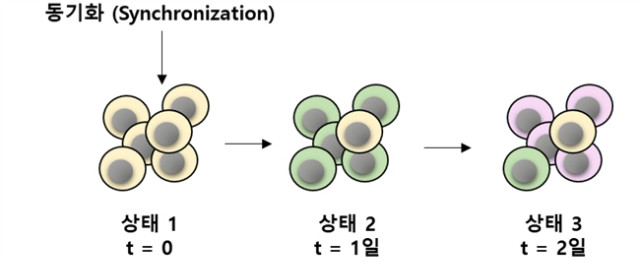
мғқл¬јн•ҷм Ғ н”„лЎңм„ёмҠӨ(biological process)лҠ” лӘҮ 분м—җм„ң лӘҮ мӢңк°„(нҷҳкІҪм Ғ мһҗк·№м—җ лҢҖн•ң л№ лҘё л°ҳмқ‘), лӘҮ мӢңк°„м—җм„ң л©°м№ (м„ёнҸ¬ 분нҷ”) л°Ҹ лӘҮ л…„(л°ңлі‘) л“ұмқҳ лҸҷм Ғмқё кё°к°„мқ„ ліҙмқёлӢӨ. м„ёнҸ¬лҠ” мҷ„лІҪн•ң лҸҷкё°нҷ”(synchrony)к°Җ мўҖмІҳлҹј м–ҙл өкі к·ёл“Өмқҳ м—ӯлҸҷм„ұмқҙ 비 кІ°м •м Ғмқҙкё° л•Ңл¬ём—җ, лҸҷм Ғ кіјм •мқҳ кё°мҙҲк°Җ лҗҳлҠ” мЎ°м Ҳ мқҙлІӨнҠёл“Өмқ„ нҠ№м„ұнҷ”н•ҳлҠ” кІғмқҖ м–ҙл Өмҡё мҲҳ мһҲлӢӨ(к·ёлҰј 1).
к·ёлҰј 1. мӢңк°„м Ғ 축. кі„мҶҚн•ҙм„ң лҸҷмӢңм„ұмқ„ мһғм–ҙлІ„лҰ¬лҠ” м„ёнҸ¬мқҳ лІҢнҒ¬ 분м„қ мғҳн”Ң 집лӢЁмқҖ мӢңк°„м Ғ м—ӯлҸҷм„ұмқҳ м •нҷ•н•ң м¶”лЎ м—җ н•ңкі„к°Җ мһҲлӢӨ.
лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷмқҖ мқҙлҹ¬н•ң н•ңкі„м җл“Өмқ„ л¶Җ분м ҒмңјлЎң мҷ„нҷ”н•ҙмӨҖлӢӨ. нҳ„мһ¬ лҢҖл¶Җ분мқҳ мң м „мІҙ кё°мҲ л“ӨмқҖ мӮҙм•„мһҲлҠ” м„ёнҸ¬ 추м Ғкіј м–‘лҰҪн• мҲҳ м—Ҷм§Җл§Ң, мӣҗм№ҷм ҒмңјлЎң 비лҸҷкё°(asynchronous) 집лӢЁм—җм„ңмқҳ м„ёнҸ¬ мғҳн”Ңл§ҒмқҖ м—°кө¬мӨ‘мқё м„ёнҸ¬ 집лӢЁм—җм„ң лӮҳнғҖлӮҳлҠ” м–ҙлҠҗ мӢңм җм—җм„ңлҸ„ м„ёнҸ¬мқҳ м—ӯн•ҷ(cellular dynamics)м—җ лҢҖн•ң кі„мӮ° лӘЁлҚёмқ„ к°ңл°ңн• мҲҳ мһҲлӢӨ. л”°лқјм„ң лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷмқҖ мң м „мІҙ кё°лҠҘ л°Ҹ мЎ°м Ҳ лӮҙмқҳ м—ӯн•ҷмқ„ м¶”лЎ н•ҳкё° мң„н•ң ліҙнҺём Ғмқҙкі кі„мӮ°м Ғмқё м ‘к·јл°©мӢқмқ„ м ңм•Ҳн•ңлӢӨ.
лӢЁмқј м„ёнҸ¬ лҚ°мқҙн„°лЎңл¶Җн„° м„ёнҸ¬мқҳ м—ӯн•ҷ м¶”лЎ мқҳ к·јк°„мқ„ мқҙлЈЁлҠ” кё°ліёмӣҗлҰ¬лҠ” мөңлҢҖмқҳ м Ҳм•Ҫ(maximum parsimony)мқҙлӢӨ. мқҙлҠ” кҙҖм°°лҗң м„ёнҸ¬мқҳ мғҒнғңлҘј м—°кІ°н•ҳлҠ” лӘЁл“ к°ҖлҠҘн•ң м—ӯн•ҷ лӘЁлҚёмқ„ м ңм•Ҳн•ҳл©°, м „мӮ¬м—җм„ң мөңмҶҢмқҳ ліҖнҷ”мқё кІғмқ„ м„ нҳён•ңлӢӨ. мқҙ мӣҗлҰ¬лҠ” мң м „мһҗ л°ңнҳ„м—җм„ң мЎ°м •лҗң ліҖнҷ”мқҳ мқјл Ёмқҳ мҙҲм җ(focal point)мқ„ нҶөн•ҙ л°©н–Ҙм„ұ л°Ҹ 비к°Җм—ӯм ҒмңјлЎң 진н–үн•ҳлҠ” м„ёнҸ¬ м—ӯн•ҷмқ„ м¶”лЎ н•ҳлҠ”лҚ° нҡЁкіјм ҒмқҙлӢӨ. мқҙлҹ¬н•ң кІҪмҡ°м—җ, м„ёнҸ¬мқҳ 'orderingвҖҷ(м„ёнҸ¬к°Җ мЎҙмһ¬н•ҳлҠ” мқҙмғҒм Ғмқё мӢңк°„м Ғ кіјм •мқҳ м§Җм җ)кіј мЈјмҡ” 분нҷ” лҳҗлҠ” 분нҷ” м§Җм җм—җм„ң м„ёнҸ¬мҷҖ кҙҖл Ёлҗң 분мһҗ мғҒнғңлҘј м¶”лЎ н• мҲҳ мһҲм–ҙм•ј н•ңлӢӨ. лҳҗн•ң, мғҳн”Ңл§Ғлҗң лӘЁл“ лӢЁмқј м„ёнҸ¬лҠ” кё°лҠҘм Ғ н”„лЎңм„ёмҠӨ(functional process)мқҳ м–ҙл”ҳк°Җм—җ мң„м№ҳн•ҳкё° л•Ңл¬ём—җ, к·ё кіјм •мқҳ нҠ№м • к°„кІ©м—җм„ңмқҳ 'мІҙлҘҳ мӢңк°„(residence time)'мқҖ мғҳн”Ңл§Ғлҗң м„ёнҸ¬мқҳ 비мңЁкіј кҙҖл Ёмқҙ мһҲлӢӨ. мғҳн”Ңл§Ғлҗң м„ёнҸ¬ мҲҳк°Җ мғҒлӢ№н•ҳл©ҙ, к·№лҸ„лЎң мқјмӢңм Ғмқё мғҒнғңмқҙлҚ”лқјлҸ„ 추측лҗң к¶Өм Ғм—җ нҷ•мӢӨнһҲ мң„м№ҳ н• мҲҳ мһҲлӢӨ. к·ёлҹ¬лҜҖлЎң мғқл¬јн•ҷм Ғ 비лҸҷкё° (biological asynchrony)лҠ” мһҗмӮ°мқҙл©°, лӢЁкё°к°„м—җ л°ҳліөм ҒмңјлЎң л°ңмғқн•ҳлҠ” кіјм •(мҳҲ. мЎ°нҳҲ; haematopoiesis)м—җ лҢҖн•ң м „мІҙ лҸҷм Ғ кіјм •мқ„ лӢЁмқј м„ёнҸ¬мқҳ л§Өмҡ° к№ҠмқҖ мғҳн”Ңл§Ғмқ„ нҶөн•ҙ н•ң лІҲм—җ нҡЁкіјм Ғмқё мғҳн”Ңл§Ғмқ„ н• мҲҳ мһҲлӢӨ.
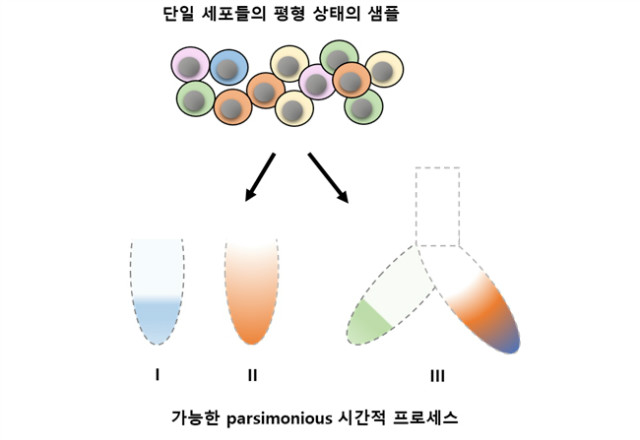 к·ёлҰј 2. мӢңк°„м Ғ м—ӯлҸҷм„ұ м¶”лЎ . лӢӨм–‘н•ң мғҒнғң лӮҙмқҳ лӢЁмқј м„ёнҸ¬л“Өмқҳ мқҙм§Ҳм Ғ нҳјн•©л¬јмқҳ мғҳн”Ңл§ҒмқҖ maximum parsimony principle кё°л°ҳмңјлЎң мӢңк°„м Ғ м—ӯлҸҷм„ұ м¶”лЎ мқ„ мң„н•ҙ мӮ¬мҡ©лҗңлӢӨ. мғҳн”Ңл§Ғлҗң м„ёнҸ¬л“ӨмқҖ м„ нҳ• 추세 (I, II) лҳҗлҠ” 분기нҳ• (III) н”„лЎңм„ёмҠӨлЎң м •л ¬лҗңлӢӨ. мқјл Ёмқҳ м—°кө¬л“ӨмқҖ нҳ„мһ¬ м„ нҳ•(linear), мЈјкё°(cyclic) лҳҗлҠ” 분기 к¶ӨлҸ„(bifurcating trajectory)лҘј к°Җм •н•ҳл©ҙм„ң, лӢЁмқј м„ёнҸ¬л“Өмқҳ н”„лЎңнҢҢмқјлЎңл¶Җн„° м—ӯн•ҷмқ„ м¶”лЎ н•ҳкё° мң„н•ҙ maximum parsimonyлҘј мқҙмҡ©н–ҲлӢӨ. мҳҲлҘј л“Өм–ҙ, Wanderlustлқјкі л¶ҲлҰ¬лҠ” мҙҲкё° л°©лІ•мқҖ B м„ёнҸ¬ 분нҷ”мқҳ к¶Өм Ғмқ„ л§Ңл“Өкё° мң„н•ҙ м§Ҳлҹү м„ёнҸ¬ мёЎм •(mass cytometry) лҚ°мқҙн„°лЎңл¶Җн„° лӢЁмқј м„ёнҸ¬ ліөн•© лӢЁл°ұм§Ҳ мёЎм •мқ„ н•ҳмҳҖлӢӨ. кҙҖл Ёлҗң м ‘к·јлІ•л“ӨмқҖ мІҙмҷё к·јмңЎнҳ•м„ұ(myogenesis in vitro) лҳҗлҠ” мІҙлӮҙ мӢ кІҪ л°ңмғқ(neurogenesis in vivo)мқҳ м—¬лҹ¬ мӢңм җм—җм„ң мғҳн”Ңл§Ғлҗң лӢЁмқј м„ёнҸ¬ RNA-seq лҚ°мқҙн„°лЎңл¶Җн„° м„ұкіөм ҒмңјлЎң м„ёнҸ¬лҘј м •л ¬н–ҲлӢӨ. м„ёнҸ¬мЈјкё°лҸҷм•Ҳ лҸҷкё°нҷ” лҗң м„ёнҸ¬ 집лӢЁмқҳ лІҢнҒ¬ н”„лЎңнҢҢмқјмқ„ 비көҗн•ҳлҠ” кІғмқҖ м–ҙл өм§Җл§Ң, лӢЁмқј м„ёнҸ¬мқҳ мЈјкё°лҠ” мүҪкІҢ мһ¬кө¬м„ұ лҗ мҲҳ мһҲлӢӨ. к°ҖмһҘ мөңк·јмқҳ л°©лІ•л“Ө лҳҗн•ң 분기лҘј мһ¬кө¬м„ұн•ҳлҠ” кІғм—җ лҢҖн•ҙ кі л¬ҙм Ғмқё м§„м „мқ„ ліҙмҳҖмңјл©°, м„ёнҸ¬мқҳ м—ӯн•ҷмқҖ нӣ„м„ұмң м „мІҙ лҚ°мқҙн„°лЎңл¶Җн„°лҸ„ м¶”лЎ лҗҳм–ҙмҷ”лӢӨ.
к·ёлҹ¬лӮҳ, мқјл Ёмқҳ мқҙлІӨнҠё лҳҗлҠ” лҸҷм Ғ кіјм •мқҳ 분기 кө¬мЎ°м—җ лҢҖн•ң мғҲлЎңмҡҙ м¶”лЎ мқҖ к№ҠкІҢ мғҳн”Ңл§Ғлҗң н”„лЎңм„ёмҠӨмқҳ кІҪмҡ°м—җлҸ„ кі„мӮ°м ҒмңјлЎң м–ҙл өлӢӨ. нҠ№нһҲ, кё°мӨҖм җ(anchor point) кІ°м •мқ„ мң„н•ң мӮ¬м „ м§ҖмӢқмқҙ кұ°мқҳ м—ҶлҠ” кІҪмҡ°, лҚ°мқҙн„°м—җ мқҳн•ҙ л¶Ҳ충분н•ң кІ°м •мқҙ мқҙлЈЁм–ҙм§Ҳ мҲҳ мһҲлӢӨ. нҠ№нһҲ maximum parsimony мӣҗм№ҷмқҙ н•ӯмғҒ м Ғм Ҳн•ң кІғмқҖ м•„лӢҲлӢӨ. мң м „мһҗмқҳ мёЎл©ҙ м „мқҙ(lateral transfer)лҘј кі л Өн•ң лӘЁлҚёмқҳ кі„нҶө л°ңмғқ мһ¬кө¬м„ұмқҳ м–ҙл ӨмӣҖкіј мң мӮ¬н•ҳкІҢ, мһ¬л¶„нҷ”(transdifferentiaion), к°ҖмҶҢм„ұ(plasticity) лҳҗлҠ” л¶Ҳмҷ„м „н•ң м„ёнҸ¬ кі„нҶө 분лҘҳлҘј мң„н•ҙм„ңлҠ” нҳ„мһ¬мқҳ parsimony кё°л°ҳмқҳ мһ¬кө¬м„ұ м•Ңкі лҰ¬мҰҳкіј мғҲлЎңмҡҙ кі„мӮ° л°Ҹ мӢӨн—ҳм Ғ м ‘к·јлІ•мқҳ нҶөн•©мқҙ мҡ”кө¬лҗңлӢӨ. кІҢлӢӨк°Җ м„ёнҸ¬лҠ” лҸҷмӢңм—җ м—¬лҹ¬ лҸҷм Ғ кіјм •(мҳҲ. мҳҒм–‘ л¬јм§Ҳм—җ л°ҳмқ‘н•ҳкі л¶„нҷ”мқҳ нҠ№м • м§Җм җм—җм„ң м„ёнҸ¬ 분м—ҙмқ„ кІӘлҠ” кІҪмҡ°)мқ„ кұ°м№ҳкё° л•Ңл¬ём—җ, мқҙлҹ¬н•ң кіјм •мқ„ м„ӨлӘ…н•ҳкі кө¬лі„н•ҳкё° мң„н•ң л°©лІ•мқҙ н•„мҡ”н•ҳлӢӨ. м¶”лЎ лҗң м—ӯн•ҷм Ғ лӘЁлҚёмқ„ нҳ•нғңн•ҷм Ғ лҳҗлҠ” мң м „м Ғ нҠ№м§•кіј к°ҷмқҖ лӢӨлҘё лҸ…лҰҪм Ғмқё мёЎм • л°©лІ•кіј м—°кІ°н•ҳлҠ” кІғмқҙ мӨ‘мҡ”н• кІғмқҙлӢӨ. мҳҲлҘј л“Өм–ҙ, B м„ёнҸ¬м—җм„ңмқҳ к¶Өм Ғ 분м„қмқ„ нҶөн•ҙ, лӘЁл“ м„ёнҸ¬мқҳ 0.007%л§ҢмңјлЎң кө¬м„ұлҗң мҙҲкё°мқҳ м¶”м • кө°м§‘ мһ„мқ„ нҷ•мқён•ҳмҳҖмқ„ л•Ң, мқҙ мғҲлЎңмҡҙ кө°м§‘мқҳ мӢңк°„м Ғ мң„м№ҳлҠ” л©ҙм—ӯ кёҖлЎңл¶ҲлҰ° мӨ‘мҮ„ мҳҒм—ӯ(immunoglobulin heavy chain locus)мқҳ мғҒнғңлҘј мЎ°мӮ¬н•ЁмңјлЎңмҚЁ нҷ•мқё к°ҖлҠҘн•ҳлӢӨ. мӢӨм ңлЎң, T м„ёнҸ¬ л°Ҹ B м„ёнҸ¬мқҳ кІҪмҡ° T м„ёнҸ¬ н•ӯмӣҗ мҲҳмҡ©мІҙ л°Ҹ B м„ёнҸ¬ мҲҳмҡ©мІҙ л©ҙм—ӯ кёҖлЎңл¶ҲлҰ°мқҳ лӢЁмқј м„ёнҸ¬ RNA-seq лҚ°мқҙн„°лҘј нҶөн•ҙ к·ёл“Өмқҳ мң м „мІҙ мғҒнғңлҘј м§Ғм ‘м ҒмңјлЎң м•Ңм•„лӮј мҲҳ мһҲлӢӨ.
лҳҗн•ң, м„ёнҸ¬мқҳ м—ӯн•ҷ м¶”лЎ мқҖ лҸҷмқј м„ёнҸ¬м—җм„ң DNAмҷҖ RNAлҘј лӘЁл‘җ мёЎм •н•ҳлҠ” кё°лІ•мқҳ 진ліҙм—җ л”°лқј нҒ¬кІҢ н–ҘмғҒ лҗ кІғмқҙлӢӨ. мғҳн”Ңл§Ғлҗң м„ёнҸ¬мҷҖ кіөнҶө мЎ°мғҒ м„ёнҸ¬(progenitor cell)лҘј кі„мёөм ҒмңјлЎң м—°кҙҖмӢңнӮӨлҠ” м„ёнҸ¬ кі„нҶө м§ҖлҸ„(cell-lineage map)лҠ” мң м „мІҙ м •ліҙлЎңл¶Җн„° м¶”лЎ к°ҖлҠҘн•ҳлӢӨ. мҳҲлҘј л“Өм–ҙ, лӢЁмқј м„ёнҸ¬ RNA-seqмқ„ нҶөн•ҙ м–»мқҖ м„ёнҸ¬мқҳ кё°лҠҘм Ғ лҸҷмқјм„ұ(functional identity)м—җ лҢҖн•ң м§ҖмӢқкіјмқҳ м—°кІ°мқ„ нҶөн•ҙ, м„ёнҸ¬-мҡҙлӘ… м§ҖлҸ„(cell-fate map, мҙҲкё°мқҳ м„ёнҸ¬ мң нҳ•мқҙ мқҙнӣ„мқҳ м„ёнҸ¬ мң нҳ•мқ„ мқјмңјнӮҙмқ„ л°қнҳҖлӮё)лҘј м–»м–ҙлӮҙлҠ” кІғмқҙ к°ҖлҠҘн• кІғмқҙлӢӨ. лӢЁмқј м„ёнҸ¬мқҳ мӢңк°„м Ғ м—ӯн•ҷмқ„ мқҙн•ҙн•ҳлҠ” кІғмқҖ мқёк°„мқҳ м§Ҳлі‘(нҠ№нһҲ м•”)мқ„ л°қнһҲлҠ”лҚ° мӨ‘мҡ”н•ҳлӢӨ. мў…м–‘ нҳ•м„ұмқҖ л§Өмҡ° м—ӯлҸҷм Ғмқҙл©° мқҙм§Ҳм Ғмқё нҷҳкІҪм Ғ мғҒнҷ©м—җм„ң мң м „мІҙ л°Ҹ нӣ„м„ұмң м „мІҙмқҳ ліҖнҷ”лҘј лӘЁл‘җ мҲҳл°ҳн•ңлӢӨ. к·ёлҹ¬лӮҳ нҳ„мһ¬мқҳ л°©лІ•мңјлЎңлҠ” нҷҳмһҗмқҳ мў…м–‘м—җм„ң лӢЁ н•ҳлӮҳ лҳҗлҠ” лӘҮ к°ңмқҳ мӢңлЈҢ л§Ңмқ„ м–»мқ„ мҲҳ мһҲлӢӨ. мў…м–‘ мғқкІҖ лӮҙмқҳ к°Ғ м•…м„ұ м„ёнҸ¬мқҳ мң м „мІҙ л°Ҹ кё°лҠҘм Ғ мғҒнғңмқҳ лҸҷмӢң мёЎм •мқҖ мў…м–‘мқҳ 진нҷ” 추м Ғ, м „мқҙмқҳ мқҙн•ҙ, м№ҳлЈҢ л°ҳмқ‘мқҳ лӘЁлӢҲн„°л§Ғ л°Ҹ мҳҲмёЎ л“ұмқ„ н• мҲҳ мһҲлҠ” лҶҖлқјмҡҙ кё°нҡҢлҘј м ңкіө н• кІғмқҙлӢӨ. мөңк·јмқҳ м—°кө¬лҠ” нқ‘мғүмў…(melanoma), көҗлӘЁм„ёнҸ¬мў…(glioblastoma), мң л°©м•”(breast cancer), нқ¬мҶҢ лҸҢкё° м•„нҳ• (oligodendroglioma) л°Ҹ л°ұнҳҲлі‘(leukaemia)м—җм„ң мқҙлҹ¬н•ң м ‘к·јлІ•мқҳ к°ҖлҠҘм„ұмқ„ ліҙм—¬мЈјм—ҲлӢӨ.
4. кіөк°„м Ғ 축мқҳ м¶”лЎ мғқлҰ¬н•ҷм Ғ н”„лЎңм„ёмҠӨл“ӨмқҖ мЎ°м§Ғ лӮҙм—җм„ң мқјм–ҙлӮҳл©°, кіөк°„м Ғ кө¬м„ұмқҖ мЎ°м§Ғмқҳ кё°лҠҘм—җ мӨ‘мҡ”н•ҳм§Җл§Ң, нҳ„мһ¬ лӢЁмқј м„ёнҸ¬ мң м „мІҙ м ‘к·јлІ•л“ӨмқҖ мЎ°м§Ғ мғҳн”Ңл“Өмқ„ н•ҙмІҙн•ҳл©°, м„ёнҸ¬мқҳ 3D кө¬м„ұ л“ұлЎқмҶҢ(registry)лҘј мң м§Җн•ҳм§Җ лӘ»н•ңлӢӨ. н•ҳм§Җл§Ң лӘҮ к°Җм§Җ н•ҙкІ°мұ…мқҙ л°ңлӢ¬лҗҳкі мһҲмңјл©°, н•ҳлӮҳмқҳ м ‘к·јлІ•мқҖ кіөк°„м Ғ н•ҙкІ° л¬ём ңлҘј кі„мӮ°м Ғ лӘЁлҚёлЎң кІ©н•ҳмӢңнӮӨлҠ” кІғмқҙлӢӨ(к·ёлҰј 2). м„ёнҸ¬мқҳ м „мӮ¬мІҙлҠ” нқ”нһҲ кі мң мқҳ мң„м№ҳлҘј м§ҖлӢҲкі мһҲлӢӨ. лӢЁмқј м„ёнҸ¬мқҳ н”„лЎңнҢҢмқјкіј лӘҮлӘҮ л§Ҳм»Ө мң м „мһҗ(marker gene)мқҳ м°ёмЎ° м§ҖлҸ„(reference map)лҘј кІ°н•©мқ„ нҶөн•ҙ, м—¬лҹ¬ м—°кө¬л“ӨмқҖ лӢЁмқј м„ёнҸ¬лҘј к·ёл“Өмқҳ кіөк°„м Ғ мң„м№ҳлЎң лҸҢл ӨлҶ“м•ҳлӢӨ. мҳҲлҘј л“Өм–ҙ, мҙҲкё° мғқм„ л°°м•„м—җ лҢҖн•ң м—°кө¬лҠ” мөңлҢҖ 100к°ңмқҳ м„ёнҸ¬л“Өмқ„ кіөк°„м Ғ 'bins'м—җ н• лӢ№н•ҳкё° мң„н•ҙ, лҸҷмқјн•ң л°ңлӢ¬ лӢЁкі„мқҳ лӢЁмқј м„ёнҸ¬ RNA-seq лҚ°мқҙн„°мҷҖ мҲҳмӢӯ к°ңмқҳ л§Ҳм»Өм—җ лҢҖн•ң in-situ л°ңнҳ„ лҚ°мқҙн„°мқҳ м°ёмЎ° м§ҖлҸ„лҘј кІ°н•©н•ҳмҳҖлӢӨ. мқҙлҠ” кіөк°„м ҒмңјлЎң лҸ…лҰҪм Ғмқё м„ёнҸ¬ мң нҳ• м§Җм • н”„лЎңм„ёмҠӨ(cell-type specification process)лЎңл¶Җн„° кө¬лі„мқ„ мң„н•ҙ кіөк°„м Ғ ліҖнҷ”мҷҖ к°•н•ҳкІҢ м—°кІ°лҗң мң м „мһҗ л°ңнҳ„ мӢңк·ёлӢҲмІҳлҘј к°ҖлҠҘн•ҳкІҢ н–ҲлӢӨ. 비мҠ·н•ң м „лһөмңјлЎң лІҢл Ҳ лҮҢм—җм„ң м җ лӘЁм–‘мқҳ нҢЁн„ҙмңјлЎң 분нҸ¬лҗң м„ёнҸ¬л“Өмқ„ л§Өн•‘н•ҳмҳҖмңјл©°, лӢӨлҘё м—°кө¬л“ӨмқҖ мҙҲкё° л§Ҳмҡ°мҠӨ л°°м•„ л°Ҹ м„ұмІҙ л§Ҳмҡ°мҠӨ н•ҙл§ҲлЎңл¶Җн„°мқҳ лӢЁмқј м„ёнҸ¬ н”„лЎңнҢҢмқјм—җм„ң лі„к°ңмқҳ мҳҒм—ӯ л°Ҹ нҳ•нғң л°ңмғқм Ғ ліҖнҷ”(morphogen gradient)лҘј ліҙмҳҖлӢӨ.
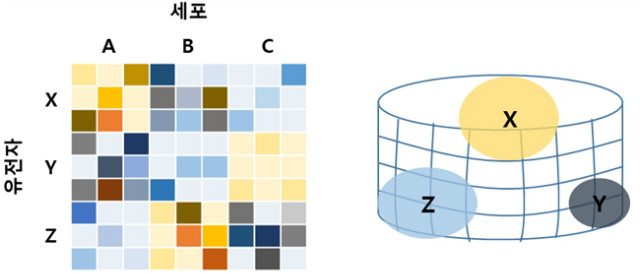
к·ёлҰј 3. кіөк°„м Ғ 축. кіөк°„м Ғ л§өн•‘мқҖ лӢЁмқј м„ёнҸ¬ мң м „мһҗ л°ңнҳ„ н”„лЎңнҢҢмқјл§Ғ(мҷјмӘҪ)кіј, мҶҢмҲҳмқҳ лһңл“ңл§ҲнҒ¬ мң м „мһҗл“Өмқҳ кіөк°„м Ғ л°ңнҳ„ нҢЁн„ҙм—җ лҢҖн•ң м°ёмЎ° м§ҖлҸ„лҘј мһ…л Ҙк°’мңјлЎң мӮ¬мҡ©н•ңлӢӨ.
мқҙлҹ¬н•ң мҳҲл“ӨмқҖ лҢҖк°•мқҳ м •ліҙм—җм„ң мӢӨн—ҳм Ғ л””мһҗмқё(мҳҲ. нҳ„лҜён•ҙл¶ҖлЎңл¶Җн„°)кіј лӢЁмқј м„ёнҸ¬ н”„лЎңнҢҢмқјкіјмқҳ кІ°н•©мқҙ кіөк°„м Ғ мң м „мІҙм—җ лҢҖн•ң н’Қл¶Җн•ң м •ліҙлҘј м ңкіөн•ҳлҠ”лҚ° лҸ„мӣҖмқҙ лҗ мҲҳ мһҲмқҢмқ„ мӢңмӮ¬н•ңлӢӨ. к·ёлҹ¬лӮҳ л°°м№ҳ нҺён–Ҙ(batch bias)мқ„ мөңмҶҢнҷ”н•ҳкі мЎ°м§Ғ м ҲнҺёл“Ө к°„мқҳ мқјкҙҖм„ұмқ„ лҶ’мқҙкё° мң„н•ҙ, лӢЁмқј м„ёнҸ¬ мң м „мІҙ л°©лІ•кіј мқҙлҹ¬н•ң н•ҙл¶Җн•ҷм Ғ л°©лІ•мқҳ нҳёнҷҳм„ұмқҖ к°ңм„ лҗҳм–ҙм•ј н•ңлӢӨ. ліҙлӢӨ к·јліём ҒмңјлЎң, нҳ„мһ¬ кө¬нҳ„лҗң кі„мӮ°м Ғ(computational) кіөк°„м Ғ мһ¬кө¬м„ұмқҖ л°°м•„ л°ңмғқ лҳҗлҠ” кё°кҙҖ нҳ•м„ұ(organogenesis)м—җ мһҲм–ҙм„ң н‘ңмӨҖ мЎ°м§Ғ кө¬м„ұмқҳ м•„мқҙл””м–ҙм—җ мқҳмЎҙн•ңлӢӨ. л”°лқјм„ң ліөн•©м Ғмқё мӢӨн—ҳл“Өмқ„ нҶөн•ҙ лҸҷмқјн•ң кө¬мЎ°лҘј м •нҷ•нһҲ мһ¬нҳ„ н• мҲҳ мһҲлӢӨ. лҚң м ңн•ңм Ғмқё л°ңлӢ¬, 분нҷ” лҳҗлҠ” лі‘лҰ¬н•ҷм Ғ мӢңлӮҳлҰ¬мҳӨм—җ лҢҖн•ң м „мӮ° 분м„қмқ„ мң„н•ҙ, мһ¬нҳ„м„ұ мһҲлҠ” кіөк°„м Ғ кө¬мЎ°к°Җ мӮ¬мҡ©лҗ мҲҳ мһҲлӢӨ. мҳҲлҘј л“Өм–ҙ, мЎ°м§Ғ лі‘лҰ¬н•ҷ(histopathology)мқҖ мһ„мғҒ лі‘лҰ¬н•ҷ (clinical pathology)мқҳ к·јкұ°лҘј нҳ•м„ұн•ҳлҠ” мғҳн”Ңл“Ө к°„м—җ ліҙмЎҙлҗҳлҠ” кі м°Ёмӣҗ нҠ№м„ұл“Өмқҙ мһҲмқҢмқ„ ліҙмҳҖлӢӨ. лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷм—җм„ң кіөк°„ 분м„қмқ„ мң„н•ң ліҙнҺём Ғмқё м „лһөмқ„ к°•кө¬н•ҳкё° мң„н•ҙм„ңлҠ”, м—¬лҹ¬ к·ңлӘЁм—җм„ң мӮ¬мҡ©н• мҲҳ мһҲлҠ” мғҲлЎӯкі мң м—°н•ң м „мӮ° л°©лІ•мқ„ кіөк°„ кө¬мЎ°лҘј м§Ғм ‘м ҒмңјлЎң мЎ°мӮ¬н•ҳлҠ” н–ҘмғҒлҗң мӢӨн—ҳ кё°лІ•кіј н•Ёк»ҳ нҶөн•©н•ҙм•ј н•ңлӢӨ.
мқҙлҹ¬н•ң л¬ём ңлҘј н•ҙкІ°н•ҳкё° мң„н•ҙ мЎ°м§Ғ м ҲнҺём—җм„ң лӢЁмқј м„ёнҸ¬мқҳ in situ мң м „мІҙ 분м„қмқ„ мң„н•ң кё°мҲ л“Өмқҙ л№ лҘҙкІҢ 진нҷ”н•ҳкі мһҲлӢӨ. MERFISHмҷҖ к°ҷмқҖ лӢӨмӨ‘ RNA нҳ•кҙ‘ in situ hybridization (FISH) кё°мҲ мқҖ мҲҳмІң к°ңмқҳ лӢӨлҘё м „мӮ¬мІҙмқҳ л°ңнҳ„ л°Ҹ кіөк°„м Ғ мң„м№ҳлҘј лҸҷмӢңм—җ м•Ҳм •м ҒмңјлЎң мёЎм •н•ңлӢӨ. лӢӨлҘё кё°мҲ мқҖ ліҙмЎҙлҗң мЎ°м§Ғ м ҲнҺёкіј м„ёнҸ¬м—җм„ң in situ RNA-seqмқ„ мң„н•ң к°ңл…җ мҰқлӘ… (proof-of-concept) лҚ°мқҙн„°лҘј л§Ңл“Өм–ҙ лғҲлӢӨ. лҳҗн•ң к°Ғ м ңкұ°лҗң 'pixel'мқҳ mass-cytometry мёЎм •кіј н•Ёк»ҳ мЎ°м§Ғмқҳ л Ҳмқҙм Җ лҳҗлҠ” мқҙмҳЁ л№” м ңкұ°мқҳ кІ°н•©мңјлЎң л©ҖнӢ°н”Ңл үмҠӨм—җм„ң мҲҳмӢӯ к°ң лӢЁл°ұм§Ҳмқҳ кіөк°„м Ғ л°ңнҳ„мқ„ м•Ңм•„лӮҙлҠ” кІғмқҙ к°ҖлҠҘн•ҳлӢӨ. мөңк·ј м—°кө¬м—җм„ңлҠ” мң л°©м•” мЎ°м§Ғмқ„ 분м„қн•ҳкі , м„ёнҸ¬ кі мң мқҳ н”„лЎңнҢҢмқјліҙлӢӨ мЈјліҖ м„ёнҸ¬м—җ мқҳн•ң мң нҳ•мқ„ 분лҘҳн•ҳкё° мң„н•ҙ imaging mass-cytometryлҘј мӮ¬мҡ©н–ҲлӢӨ. мқҙлҹ¬н•ң л°©лІ•л“Өмқ„ мӮ¬мҡ©н• л•Ң, м„ёнҸ¬ мң нҳ•мқҳ к°ңл…җмқ„ кіөк°„м Ғ мғҒнҷ©мқ„ нҸ¬н•Ён•ҳлҸ„лЎқ мқјл°ҳнҷ”н• мҲҳ мһҲмңјл©°, нҳјн•©лҗң мң м „мһҗ мЎ°м Ҳ л©”м»ӨлӢҲмҰҳкіј н”„лЎңк·ёлһЁмқ„ кіөк°„м Ғ нҠ№м§•мңјлЎң мҷ„м „нһҲ мһ¬м •мқҳ н• мҲҳлҸ„ мһҲлӢӨ.
мЎ°м§Ғмқҳ кіөк°„м Ғ кө¬мЎ°мқҳ мӨ‘мҡ”н•ң мқҳлҜёлҠ” нҠ№м • м„ёнҸ¬ мң нҳ•мқҳ мғҒлҢҖм Ғмқё мң„м№ҳмҷҖ к·ёкІғл“Ө мӮ¬мқҙмқҳ л¬јлҰ¬м Ғ м ‘мҙүмқҳ 분мһҗм Ғ кө¬м„ұ(molecular composition)мқҙлӢӨ. м§Ғм ‘м Ғмқё м„ёнҸ¬мқҳ м ‘мҙүмқҖ м„ёнҸ¬ к°„ мЎ°м Ҳ л©”м»ӨлӢҲмҰҳмқ„ лӘЁлҚёл§Ғ н• л•Ң нҠ№нһҲ мӨ‘мҡ”н•ҳлӢӨ. мқҙлЎ м ҒмңјлЎң, мЎ°м§Ғмқҳ мҷ„м „н•ң 3D м§ҖлҸ„лҠ” лӘЁл“ м„ёнҸ¬-м„ёнҸ¬ м ‘н•©(cell-cell junction)мқ„ нҠ№м„ұнҷ”н• мҲҳ мһҲм§Җл§Ң, м ‘к·јлІ•л“Ө лҳҗн•ң мқҙл“Өмқ„ м§Ғм ‘ мёЎм •н• мҲҳ мһҲкІҢ л°ңм „лҗҳм–ҙ мҷ”лӢӨ(мҳҲ, л‘җ м„ёнҸ¬мқҳ мһ‘кі лӢЁлӢЁнһҲ кІ°н•©лҗң кіЁмҲҳмқҳ micronichesлҘј нҸүк°Җ). кіөк°„м Ғ мЎ°м§Ғ(organization) л¬ём ңлҘј м„ёнҸ¬ мң нҳ• к°„мқҳ м ‘мҙүмқҳ 분нҸ¬лЎң м ңн•ң н• л•Ң, л§ҺмқҖ н‘ңліём—җм„ң нҠ№м • м„ёнҸ¬ мң нҳ•мқҳ мЎҙмһ¬мҷҖ 비мңЁ мӮ¬мқҙмқҳ мғҒкҙҖкҙҖкі„лҘј 분м„қн•ҳкё° мң„н•ң кі„мӮ°м Ғ м „лһөмқ„ к°•кө¬н• мҲҳ мһҲмңјл©°, мқҙм—җ лҢҖн•ң кІ°кіјлҠ” ліөмһЎн•ң нӢҲмғҲ (niche)м—җм„ң мғҒнҳёмқҳмЎҙм„ұкіј м„ёнҸ¬мқҳ мғҒнҳё мһ‘мҡ©м—җ кҙҖн•ң к°Җм„Өмқ„ мғқм„ұн•ҳлҠ”лҚ° мӮ¬мҡ©лҗ мҲҳ мһҲлӢӨ.
5. л©”м№ҙлӢҲмҰҳ 축과 мң м „мһҗ мЎ°м Ҳ лӘЁлҚёл§Ғ
нҳ„мғҒн•ҷмқ„ лӣ°м–ҙ л„ҳм–ҙ, м„ёнҸ¬мқҳ н–үлҸҷ(cellular behavior)мқҳ л ҲнҚјнҶ лҰ¬мқҳ кё°мҙҲк°Җ лҗҳлҠ” мЎ°м Ҳ л©”м»ӨлӢҲмҰҳмқ„ лӘЁлҚёл§Ғн•ҳкё° мң„н•ң мғҲлЎңмҡҙ м ‘к·јмқҙ н•„мҡ”н•ҳлӢӨ. 집мӨ‘м Ғмқё м—°кө¬м—җлҸ„ л¶Ҳкө¬н•ҳкі , к·ёлҹ¬н•ң л©”м»ӨлӢҲмҰҳмқҳ мІҙкі„м Ғмқё 분м„қмқҖ м—¬м „нһҲ мғҒлӢ№н•ң лҸ„м „ кіјм ңлЎң лӮЁм•„мһҲлӢӨ. лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷмқҖ м„ёнҸ¬ м•Ҳкіј л°–мқҳ мЎ°м Ҳ л©”м№ҙлӢҲмҰҳм—җ лӘЁлҚёл§Ғ м¶”лЎ мқ„ мң„н•ң кҙҖмёЎм Ғ(observational), кё°кі„лЎ м Ғ(mechanistic) л°Ҹ м„ӯлҸҷм Ғ (perturbational) м ‘к·јмқ„ кІ°н•©н• мҲҳ мһҲлҠ” мғҲлЎңмҡҙ кё°нҡҢлҘј м ңкіөн•ңлӢӨ. лҚ°мқҙн„° ліём—°мқҳ м„ёнҸ¬мқҳ н•ҙмғҒлҸ„ (cellular resolution)лҘј кі л Өн•ҙліј л•Ң, мҙҲкё° м—°кө¬л“ӨмқҖ м„ёнҸ¬ лӮҙ(intracellular) лӢЁкі„м—җ мҙҲм җмқ„ л§һ추м—Ҳмңјл©°, м „мӮ¬ мЎ°м Ҳм—җ көӯн•ңлҗҳм—ҲмңјлӮҳ, мқҙнӣ„ м—°кө¬л“ӨмқҖ м•„л§ҲлҸ„ м„ёнҸ¬ к°„(intercellular) л°Ҹ мЎ°м§Ғ лӢЁкі„м—җм„ңмқҳ нҶөн•©м—җ мӨ‘м җмқ„ л‘ҳ кІғмқҙлӢӨ.
мң м „мһҗ мЎ°м Ҳ л©”м»ӨлӢҲмҰҳмқ„ м¶”лЎ н•ҳлҠ” кҙҖмёЎм Ғ м ‘к·јлІ•мқҖ, кі м „м ҒмңјлЎң лӢӨм–‘н•ң мқјл Ёмқҳ лІҢнҒ¬ мғҳн”Ңл“Ө(мҳҲ, м„ңлЎң лӢӨлҘё мЎ°м§Ғ мң нҳ•, м„ёнҸ¬ мң нҳ• лҳҗлҠ” мһҗк·№л“Ө)мқҳ mRNA лӢЁкі„мқҳ 분мһҗм Ғ н”„лЎңнҢҢмқј лҳҗлҠ” нӣ„м„ұ н‘ңмӢқл“Ө мӮ¬мқҙмқҳ мғҒкҙҖм„ұ м¶”лЎ мқ„ нҶөн•ҙ м Ғмҡ©лҗҳм—ҲлӢӨ. мқҙлҹ¬н•ң 분м„қл“ӨмқҖ к°„м ‘м Ғмқё мғҒкҙҖкҙҖкі„лҘј л°ңмғқмӢңнӮӨкұ°лӮҳ лӢӨлҘё мӨ‘мҡ”н•ң мЎ°м Ҳ мғҒнҳёмһ‘мҡ©мқ„ мҲЁкёё мҲҳ мһҲкё° л•Ңл¬ём—җ, м„ёнҸ¬ м•ҷмғҒлё” н”„лЎңнҢҢмқјмқҳ нҳјн•©лҗң мғҒнғңм—җ мқҳн•ҙ нҳјлҸҲ(confound)лҗңлӢӨ. л°ҳл©ҙм—җ, лӢЁмқј м„ёнҸ¬ 분м„қмқҖ м„ёнҸ¬ лӮҙ мҲҳмӨҖмқҳ кіөліҖлҹү(covariation) 분м„қм—җ лҢҖн•ң лҚ” л§ҺмқҖ нһҳмқ„ к°Җ진лӢӨ. к°ҖмһҘ кё°ліём Ғмқё м ‘к·јлІ•мқҖ лҢҖлҹүмқҳ лӢЁмқј м„ёнҸ¬ н”„лЎңнҢҢмқјмқ„ мҲҳн–үн•ҳм—¬, лӢӨм–‘н•ң м „мӮ¬ мғҒнғңлҘј нҸ¬м°©н•ҳкі мң м „мһҗ-мң м „мһҗ мғҒкҙҖкҙҖкі„ кі„мӮ°мқ„ нҶөн•ҙ нӣ„ліҙ мЎ°м Ҳ мғҒнҳёкҙҖкі„лҘј м–»м–ҙлӮҙлҠ” кІғмқҙлӢӨ. л”°лқјм„ң мқҙлҹ¬н•ң 분м„қл“ӨмқҖ, м„ёнҸ¬ мң нҳ•л“Өмқҳ м „мӮ¬м Ғ мӢңк·ёлӢҲмІҳ(tranional signature)лҘј м •мқҳн•ҳлҠ” мң м „мһҗл“Өмқҳ н”„лЎңнҢҢмқјкіј л°ңнҳ„ нҢЁн„ҙ мӮ¬мқҙмқҳ мғҒкҙҖкҙҖкі„лҘј нҶөн•ҙ, м„ёнҸ¬ мң нҳ•мқҳ мЎ°м Ҳ мқёмһҗ(regulator)лҘј л°қнҳҖлӮёлӢӨ. кі н•ҙмғҒлҸ„мқҳ лӢЁмқј м„ёнҸ¬ 분м„қмқҖ м„ёнҸ¬ мң нҳ• л°Ҹ м•„нҳ•(subtype)мқҳ м •мқҳлҘј к°ңм„ н•ҳкі , мһ‘мқҖ м„ёнҸ¬ нӢҲмғҲ лӮҙмқҳ мғҒкҙҖ кҙҖкі„мқҳ нҷ•мқёмқ„ к°ҖлҠҘн•ҳкІҢ н•ҳм—¬, к°Җм§ңлЎң м¶”м •лҗң мң м „мһҗ-мң м „мһҗ мғҒнҳёмһ‘мҡ©мқ„ м җм°Ём ҒмңјлЎң м ңмҷён• мҲҳ мһҲкІҢ н•ңлӢӨ. мқҙлҹ¬н•ң 분м„қмқҖ л©ҙм—ӯ м„ёнҸ¬, мғҒн”ј м„ёнҸ¬ л°Ҹ лүҙлҹ°мқҳ м„ёнҸ¬ мң нҳ•мқ„ мЎ°м Ҳ (control)н•ҳлҠ” мЎ°м Ҳ мқёмһҗлҘј мҳҲмёЎн•ҳлҠ”лҚ° лҸ„мӣҖмқҙ лҗңлӢӨ.
к·ёлҹ¬лӮҳ лӢЁмқј м„ёнҸ¬ мҲҳмӨҖм—җм„ңлҸ„ мғҒкҙҖкҙҖкі„лҠ” мқёкіјкҙҖкі„(causation)лҘј мқҳлҜён•ҳм§Җ м•Ҡмңјл©°, мң м „мһҗ мЎ°м Ҳмқҳ мҳҲмёЎ лӘЁлҚёл§ҒмқҖ м„ӯлҸҷ(perturbation) мӢңмҠӨн…ңкіј кё°кі„лЎ м Ғ м ңм•Ҫ мЎ°кұҙмқҳ нҶөн•©м—җ мқҳмЎҙн•ҙм•ј н•ңлӢӨ. кІүліҙкё°м—җ к· м§Ҳн•ң 집лӢЁм—җм„ңлҸ„, мҷём Ғмқё л…ёмқҙмҰҲ лҳҗлҠ” 비лҸҷкё° л°ҳмқ‘мңјлЎң мқён•ҙ м…Җк°„ кі мң мқҳ нҺём°Ёк°Җ мһҲкё° л•Ңл¬ём—җ, к°Ғ м…Җмқ„ мһҗмІҙ м„ӯлҸҷ мӢңмҠӨн…ңмңјлЎң ліј мҲҳ мһҲлӢӨ. мқҙлҹ¬н•ң кІҪмҡ°, мғҒкҙҖкҙҖкі„мҷҖ к°ҷмқҖ кҙҖмёЎм Ғ м „лһөмқҖ мқҙлЎ м ҒмңјлЎң к°ҖлҠҘн•ҳм§Җл§Ң, мӢӨм ңм ҒмңјлЎңлҠ” мқёкіјм¶”лЎ мқ„ м§Җм§Җн•ңлӢӨ. мҳҲлҘј л“Өм–ҙ, мҙҲкё° м—°кө¬лҠ” лҸҷмӢңм—җ л°ңнҳ„н•ҳлҠ” н•ӯ-л°”мқҙлҹ¬мҠӨ мң м „мһҗ лӘЁл“Ҳмқ„ нҡҢмҲҳн•ҳкі мЎ°м Ҳ мқёмһҗлЎңм„ң STAT2мҷҖ IRF7 лӢЁл°ұм§Ҳмқ„ м •нҷ•нһҲ м—°кҙҖ짓기 мң„н•ҙ, м§Җм§ҲлӢӨлӢ№лҘҳ(lipopolysaccharide)мҷҖ н•Ёк»ҳ мһҗк·№н•ң мқҙнӣ„м—җ 15к°ңмқҳ мҲҳм§ҖмғҒм„ёнҸ¬(dendritic cell)л“Өмқҳ кіөліҖ 분м„қмқ„ мӮ¬мҡ©н–ҲлӢӨ. 비мҠ·н•ң м ‘к·јлІ•мңјлЎң м—¬лҹ¬ мҲҳм§ҖмғҒм„ёнҸ¬ м•„нҳ•м—җм„ң м§Җм§ҲлӢӨлӢ№лҘҳм—җ лҢҖн•ң in vivo л°ҳмқ‘мқҳ мЎ°м ҲмһҗлЎң IRF7мқ„ л°ңкІ¬н•ҳмҳҖлӢӨ.
м•Ҳм •м Ғмқё мғҒнғңлҘј нҸ¬н•Ён•ң мӢңмҠӨн…ңл“Өм—җм„ң н•ҳлӮҳмқҳ к°ңмІҙкө° лӮҙмқҳ м„ёнҸ¬л“ӨмқҖ лӘҮлӘҮ н‘ңнҳ„нҳ• ліҖмқҙм—җ мғҒмқ‘н•ҳлҠ” мҶҢмҲҳмқҳ к°ңлі„ лӘЁл“ңліҙлӢӨлҠ”, ліём§Ҳм Ғмқё м „мӮ¬ мғҒнғңмқҳ мҠӨнҺҷнҠёлҹјм—җ кұёміҗ мһҲмқ„ кІғмқҙлӢӨ. мҳҲлҘј л“Өм–ҙ, м„ёнҸ¬мЈјкё° мЎ°м Ҳ мң м „мһҗ лӘЁл“ҲмқҖ м„ёнҸ¬ 분нҷ” м ңм–ҙ н”„лЎңк·ёлһЁмңјлЎңл¶Җн„° лі„лҸ„лЎң л°қнҳҖм§Җкұ°лӮҳ лӘЁлҚёл§Ғ лҗ мҲҳ мһҲлӢӨ. лӢЁмқј м„ёнҸ¬ н”„лЎңнҢҢмқјл§ҒмқҖ, IL-17мқ„ л°ңнҳ„мӢңнӮӨлҠ” T-helper м„ёнҸ¬мқҳ лі‘мӣҗм„ұ(pathogenicity) л°Ҹ мһҗк°Җл©ҙм—ӯмӣҗм„ұ(autoimmunogenic) мһ мһ¬л Ҙм—җ мғҒмқ‘н•ҳлҠ” м „мӮ¬ мғҒнғң мҠӨнҺҷнҠёлҹјмқ„ нҡҢмҲҳн•ҳкі , мҠӨнҺҷнҠёлҹјкіј м—°кҙҖлҗң мң м „мһҗ лӘЁл“Ҳ л°Ҹ м¶”м • мЎ°м Ҳ мқёмһҗлҘј л°ңкөҙн•ҳлҠ” лҚ°м—җлҸ„ мӮ¬мҡ©лҗҳм—ҲлӢӨ. лӢЁмқј м„ёнҸ¬ лҚ°мқҙн„°лЎңл¶Җн„° мЎ°м Ҳ лӘЁлҚё м¶”лЎ мқ„ мң„н•ҙ мӮ¬мҡ©лҗң кё°мҲ мқҳ кі„мӮ°м Ғмқё м •көҗн•ЁмқҖ лҚ°мқҙн„° м…Ӣ(set)мқҳ нҒ¬кё°мҷҖ нҢҗлҸ…мқҳ мӢ лў°м„ұм—җ мқҳн•ң н•ңкі„к°Җ мһҲлӢӨ. лҢҖлҹүмқҳ лӢЁмқј м„ёнҸ¬л“Өмқҙ mass cytometry лҚ°мқҙн„°м—җм„ң ліҙлӢӨ м •лҹүм Ғмқё лӘЁлҚёл§Ғмқ„ м§Җм§Җн•Ёмқ„ ліҙмҳҖмңјл©°, мқҙлҠ” мқҙлҹ¬н•ң м ‘к·јлІ•мқҙ кі§ лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷм—җ м Ғмҡ©лҗ мҲҳ мһҲмқҢмқ„ мӢңмӮ¬н•ңлӢӨ.
мӢңк°„м Ғ н•ҙмғҒлҸ„(temporal resolution)лҠ” кҙҖмёЎ мһҗлЈҢлЎңл¶Җн„° мЎ°м Ҳ л©”м»ӨлӢҲмҰҳмқ„ м¶”лЎ н•ҳлҠ” лҚ° мӨ‘мҡ”н•ң м—ӯн• мқ„ н• мҲҳ мһҲлӢӨ. м „мӮ¬ліҙлӢӨ кёҙ мӢңк°„ лҸҷм•Ҳ л°ңмғқн•ҳлҠ” лҸҷм Ғ н”„лЎңм„ёмҠӨмқҳ кІҪмҡ°(мҳҲ. м„ёнҸ¬ мң нҳ• нҠ№мқҙ м „мӮ¬ мқёмһҗмқҳ м•Ҳм •м Ғмқё мң лҸ„), мӢңк°„м Ғ к¶Өм Ғмқ„ л”°лқј м •л ¬лҗң лҚ°мқҙн„° м…Ӣмқ„ мӮ¬мҡ©н•ҳм—¬, нҠ№м • мқҙн–ү(transition)кіј кҙҖл Ёлҗң мҙҲкё° л°Ҹ нӣ„кё° мЎ°м Ҳмһҗмқҳ нҷңлҸҷ н”„лЎңнҢҢмқј к°„мқҳ мӢңк°„ м§Җм—°мқ„ нҷ•мқён•ЁмңјлЎңмҚЁ мқёкіј м¶”лЎ мқҳ нһҳмқ„ мғҒлӢ№нһҲ н–ҘмғҒмӢңнӮ¬ мҲҳ мһҲлӢӨ. мқҙ м ‘к·јлІ•мқҖ мғқмІҙ лӮҙ мҙҲкё° B м„ёнҸ¬ л°ңлӢ¬м—җм„ң IL7/STAT5 кІҪлЎңмқҳ м—ӯн• , in vitro к·јм•„м„ёнҸ¬ 분нҷ”(myoblast differentiation)мқҳ лӢӨм–‘н•ң м „мӮ¬ мЎ°м Ҳмһҗ, л°°м•„ л°ңлӢ¬ лҸҷм•Ҳ к·ёлҰ¬кі м„ұмІҙм—җм„ңмқҳ мӢ кІҪ л°ңмғқ лҢҖн•ң мҳҲмёЎ л°Ҹ кІҖмҰқмқ„ н•ҳлҠ”лҚ° м Ғмҡ©лҗҳм—ҲлӢӨ. к·ёлҹ¬лӮҳ л§ҺмқҖ мЎ°м Ҳ н”„лЎңм„ёмҠӨл“ӨмқҖ нӣЁм”¬ 짧мқҖ мӢңк°„м—җ кұёміҗ л°ңмғқн•ҳкұ°лӮҳ м „мӮ¬ мҲҳмӨҖм—җм„ң кҙҖм°°лҗҳм§Җ м•ҠлҠ” м „мӮ¬ нӣ„ л°Ҹ лІҲм—ӯ л©”м»ӨлӢҲмҰҳмқ„ мҲҳл°ҳн•ңлӢӨ. л•Ңл¬ём—җ мӢңк°„ лӢЁмң„мқҳ(time-resolved) лӢЁмқј м„ёнҸ¬ лҚ°мқҙн„° м…Ӣм—җм„ңлҸ„ мЎ°м Ҳ кҙҖкі„л“Өмқ„ м¶”лЎ н•ҳкё° мң„н•ң лҠҘл ҘмқҖ м ңн•ңм ҒмқҙлӢӨ.
нӣ„м„ұмң м „мІҙ лҚ°мқҙн„°, нҠ№нһҲ лӢЁмқј м„ёнҸ¬ мҲҳмӨҖм—җм„ңмқҳ мң м „мһҗ мЎ°м Ҳ лӘЁлҚёмқҳ нҶөн•©мқҖ м „мӮ¬ мғҒнғңмқҳ кҙҖмёЎм—җм„ң лӮҳнғҖлӮҳм§Җ м•ҠлҠ” л¶Җ분мқ„ лҚ”н•ЁмңјлЎңмҚЁ мқёкіј м¶”лЎ мқҳ нһҳмқ„ мҰқк°ҖмӢңнӮ¬ мҲҳ мһҲлӢӨ. к°ңмІҙкө° л°Ҹ м°ёмЎ° нӣ„м„ұмң м „мІҙлҠ” м „мӮ¬ мқёмһҗлҘј кІ°н•© мҳҒм—ӯ(binding site), лҳҗлҠ” н‘ңм Ғ мң м „мһҗлҘј к°–лҠ” мқён•ём„ң(enhancer) мҡ”мҶҢмҷҖ м—°кІ°мӢңнӮҙмңјлЎңмҚЁ, мһ мһ¬м Ғ мЎ°м Ҳ мғҒнҳёмһ‘мҡ©мқҳ м ңн•ңм Ғмқё л°ңкІ¬м—җ лҸ„мӣҖмқҙ лҗ кІғмқҙлӢӨ. RNA н”„лЎңнҢҢмқјл§Ғкіј лҸ…лҰҪм Ғ лҳҗлҠ” лҸҷмӢңм—җ мҲҳн–үлҗҳлҠ” лӢЁмқј м„ёнҸ¬ нӣ„м„ұмң м „мІҙн•ҷмқҖ, нҠ№м • мЎ°м Ҳ мҡ”мҶҢм—җм„ң нӣ„м„ұм Ғ нҷңлҸҷмқ„ лӢӨлҘё мҡ”мҶҢмқҳ нҷңлҸҷ лҳҗлҠ” н‘ңм Ғ мң м „мһҗмқҳ RNA м–‘кіј м—°кІ°н•ҳм—¬, лҚ” лӮҳмқҖ н•ҙмғҒлҸ„мқҳ мң м „мІҙм—җм„ң мЎ°м Ҳ нҷҳкІҪмқ„ м •мқҳ н• мҲҳ мһҲлӢӨ. мқҙлҹ¬н•ң нӣ„м„ұм Ғ нҷңлҸҷмқҖ м—јмғүмІҙ м ‘к·јм„ұ лҳҗлҠ” DNA н•ҳмқҙнҸ¬л©”нӢёнҷ” (hy-pomethylation)лҘј нҸүк°Җн•ҳлҠ” л°©лІ•мңјлЎң нҷ•мқён• мҲҳ мһҲлӢӨ. лӢЁмқј м„ёнҸ¬ нӣ„м„ұмң м „мІҙлЎңл¶Җн„° мң м „мһҗ мЎ°м Ҳм—җ лҢҖн•ң м¶”лЎ мқҖ м—¬м „нһҲ лҚ°мқҙн„°мқҳ к№ҠмқҙмҷҖ нҸӯ, к·ёлҰ¬кі м „мӮ° м ‘к·ј л°©мӢқм—җ мқҳн•ҙ м ңн•ңлҗңлӢӨ. нғ„нғ„н•ң (robust) мғҒкҙҖкҙҖкі„ кі„мӮ°мқ„ мң„н•ң лӢӨмҲҳмқҳ нӣ„м„ұмң м „мІҙ н”„лЎңнҢҢмқјмқ„ нҶөн•©(pooling)н•ҳлҠ” л°©лІ•мқҙ мқҙнӣ„м—җ к°ңл°ңлҗҳм–ҙм•ј н•ҳл©°, мқҙлҹ¬н•ң м ‘к·јлІ•мқҖ м „мӮ¬мқҳ м •лҹүм Ғ, кё°кі„м Ғ лӘЁлҚёмқ„ к°ңл°ңн•ҳкі , м „мӮ¬ лӮҙмқҳ ліҖлҸҷм„ұкіј л…ёмқҙмҰҲлҘј мң лҸ„н•ҳлҠ” лӮҙмҷём Ғ мҡ”мқёмқҳ 분мһҗм Ғ к·јкұ°лҘј л°қнһҲлҠ”лҚ° лҸ„мӣҖмқҙ лҗ мҲҳ мһҲмқ„ кІғмқҙлӢӨ.
лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷкіј мӢӨн—ҳм Ғ м„ӯлҸҷмқҳ кІ°н•©мқҖ мқёкіј м¶”лЎ мқ„ мң„н•ң к°ҖмһҘ м§Ғм ‘м Ғмқё л°©лІ•мқ„ м ңкіөн•ңлӢӨ. ex vivo лҳҗлҠ” in vivo м—җм„ң кі м „м Ғмқё л…№м•„мӣғ(knockout) лӘЁлҚёмқҳ 분м„қмқҖ мқёмһҗл“Ө(factors) к°„мқҳ мЎ°м Ҳ мғҒнҳёмһ‘мҡ©мқҳ кІҖмҰқ л°Ҹ к°ңм„ мқ„ к°ҖлҠҘн•ҳкІҢ н–ҲлӢӨ(мҳҲ, кіЁмҲҳ м„ёнҸ¬ 분нҷ” лҸҷм•Ҳмқҳ CCAAT/мқён•ём„ң кІ°н•© лӢЁл°ұм§Ҳ C/EBPОұмҷҖ C/EBPОө). нҳ„лҢҖмқҳ лҶ’мқҖ мІҳлҰ¬лҹү(high-throughput) м„ӯлҸҷ л°©лІ•, нҠ№нһҲ CRISPR (clustered regularly interspaced short palindromic repeat) кё°мҲ мқ„ кё°л°ҳн•ң л°©лІ•л“ӨмқҖ лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷкіј кІ°н•©н•ҳм—¬ м „лЎҖ м—ҶлҠ” к·ңлӘЁмҷҖ н•ҙмғҒлҸ„лЎң мқёкіј 분м„қмқ„ мҲҳн–ү н• мҲҳ мһҲлӢӨ. мқҙлҠ” кё°мЎҙмқҳ CRISPR мҠӨнҒ¬лҰ°мқ„ лӢЁмқј м„ёнҸ¬ RNA-seq нҢҗлҸ…кіјмқҳ кІ°н•©мқ„ нҶөн•ҙ лӢ¬м„ұлҗ мҲҳ мһҲмңјл©°, лӢЁмқј л°ңнҳ„ л§Ҳм»Өмқҳ м Җн•Ёлҹү нҢҗлҸ…(low-content readout) к°’ лҢҖмӢ м—җ, л°ңнҳ„ н”„лЎңнҢҢмқјм—җ мқҳн•ҙ м ңкіөлҗҳлҠ” кі н•Ёлҹү н‘ңнҳ„нҳ•(high-content phenotype)мқ„ мӮ¬мҡ©н•ҳм—¬ м„ӯлҸҷм—җ лҢҖн•ң 분мһҗм Ғ л°ҳмқ‘мқҳ л°Җм ‘н•ң 분м„қмқ„ м ңкіөн•ңлӢӨ. мқҙлҹ¬н•ң кІ°н•©мқҖ лӢЁмқј м„ёнҸ¬ RNA-seq кё°мҲ лЎңл¶Җн„° лҢҖлҹүмқҳ мІҳлҰ¬лҹүмқ„ н•„мҡ”лЎң н•ҳл©°, 분м„қлҗң лӢЁмқј м„ёнҸ¬ к°Ғк°ҒмңјлЎңл¶Җн„° н•ҳлӮҳ мқҙмғҒмқҳ м„ӯлҸҷмқ„ нҢҗлҸ…н•ҳлҠ” м „лһөмқ„ н•„мҡ”лЎң н•ңлӢӨ. лҳҗн•ң м„ӯлҸҷ мҠӨнҒ¬лҰ°мқҖ мЎ°м Ҳ мӢңмҠӨн…ңмқ„ көҗлһҖн•ҳкі лӢЁмқј м„ёнҸ¬ мң м „мһҗ мЎ°м Ҳмқҳ кё°кі„лЎ м Ғ лӘЁлҚём—җ мқҳн•ҙ мң лҸ„лҗң к°Җм„Өмқ„ кІҖмҰқн•ҳкё° мң„н•ҙ м„Өкі„лҗ мҲҳ мһҲлӢӨ.
6. кҙҖм җ
мқёк°„ л°Ҹ лӘЁлҚё мғқл¬јмІҙм—җм„ңмқҳ м„ёнҸ¬ н”„лЎңк·ёлһЁмқҳ л§Өн•‘ л°Ҹ 분лҘҳм—җ лҢҖн•ң л…ёл ҘмқҖ, мһҘкё°(organ) л°Ҹ м „мІҙ мң кё°мІҙ лӮҙмқҳ м„ёнҸ¬ мң нҳ• л°Ҹ м•„нҳ•мқҳ нҸ¬кҙ„м Ғмқё м§ҖлҸ„лҘј м ңкіөн•ҳкё° мң„н•ҙ м җм җ лҚ” мҰқк°Җн•ҳкі мһҲлӢӨ. мқҙлҠ” м„ёнҸ¬ мң нҳ•кіј мғҒнғңм—җ лҢҖн•ң кё°мҲ м Ғ м—°кө¬лҘј л„ҳм–ҙ мЎ°м Ҳ н”„лЎңк·ёлһЁмқҳ кё°кі„лЎ м Ғ мҳҲмёЎ лӘЁлҚёмқ„ к°ңл°ңн• мҲҳ мһҲлҠ” лҶҖлқјмҡҙ кё°нҡҢлҘј м—ҙм–ҙмӨҖлӢӨ. м°ёмЎ° м§ҖлҸ„лҠ” мҳҲмёЎ лӘЁлҚёмқҳ м¶”лЎ кіј н…ҢмҠӨнҠёлҘј мң„н•ң н•„мҲҳ м¶ңл°ңм җмқҙлӢӨ. кё°кі„лЎ м Ғ лӘЁлҚёмқҖ м°ЁлЎҖлЎң нҒ° м°ёмЎ° м§ҖлҸ„ 분м„қн•ҳкі , мЈјм„қмқ„ лӢ¬кі , мқҙн•ҙн• мҲҳ мһҲкІҢ н•ңлӢӨ. л”°лқјм„ң мқёк°„мқҙлӮҳ мҘҗмқҳ м •мғҒ м„ёнҸ¬ мғҒнғңмқҳ нҸ¬кҙ„м Ғмқё м§ҖлҸ„лҘј мң„н•ҙ кё°кі„лЎ м Ғ лӘЁлҚёмқҙ н•„мҡ”н•ҳлӢӨ. к°Ғ м§Ҳлі‘мқҖ м°ёмЎ° мғҒнғңм—җм„ң мғҲлЎңмҡҙ ліҖмқҙлҘј м•јкё°н• мҲҳ мһҲкі , к°Ғ мң м „мһҗ ліҖмқҙк°Җ к·ёкІғмқ„ лҚ” ліҖнҳ•мӢңнӮӨкё° л•Ңл¬ём—җ, лӢЁмқј м„ёнҸ¬ м§ҖлҸ„(atlas)лҠ” м„ёнҸ¬ 집лӢЁм—җм„ң мғҒнғңлҘј кі„мӮ°н•ҳкё° мң„н•ң кё°мҙҲк°Җ лҗ мҲҳ мһҲлӢӨ. кҙ‘лІ”мң„н•ң м°ёмЎ° м§ҖлҸ„мқҳ мғҒнҷ©м—җм„ң, мң м „мһҗ мЎ°м Ҳ лӘЁлҚёл§ҒмқҖ м„ңм—ҙ лҳҗлҠ” нӣ„м„ұмң м „мІҙлЎңл¶Җн„° мғҒнғңлҘј мҷ„м „нһҲ мғҲлЎӯкІҢ м¶”лЎ н• н•„мҡ”лҠ” м—ҶлӢӨ. лҢҖмӢ л§Өмҡ° мң мӮ¬н•ң нҠ№м„ұмңјлЎң мёЎм •лҗң мғҒнғңлҘј мқҙмҡ©н•ҳкё° мң„н•ҙ, мһ‘мқҖ м„ӯлҸҷмқҳ мҳҒн–Ҙмқ„ м¶”лЎ н•ҳлҠ” кІғм—җ мҙҲм җмқ„ л§һ추м–ҙм•ј н•ңлӢӨ. л”°лқјм„ң м•Ңл Ө진 м„ёнҸ¬ мғҒнғңк°Җ м–ҙл–»кІҢ м„ӯлҸҷ лҗҳлҠ”м§Җ лӘЁлҚёл§Ғн•ҳлҠ” кІғмқҖ, м§Ҳлі‘мқҳ кё°м „мқ„ л°қнһҲл©ҙм„ң м„ёнҸ¬ кё°лҠҘмқҳ мҳҲмёЎ лӘЁлҚёмқ„ лӢӨлЈЁкё° мҡ©мқҙн•ң кІҪлЎңлҘј м ңкіөн•ңлӢӨ.
лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷмқҙ м„ёнҸ¬ мғҒнғңлҘј л§өн•‘н•ҳкі м„ёнҸ¬ лӮҙ мң м „мһҗ мЎ°м Ҳ л©”м»ӨлӢҲмҰҳмқ„ м¶”лЎ н•ҳлҠ” мҡ°лҰ¬мқҳ лҠҘл Ҙм—җ нҳҒлӘ…мқ„ мқјмңјнӮӨкі мһҲкё° л•Ңл¬ём—җ, лҳҗ лӢӨлҘё мӨ‘мҡ”н•ң лҸ„м „мқҖ к°ңлі„ м„ёнҸ¬ мғҒнғңлҘј м–ҙл–»кІҢ кё°лҠҘн•ҳлҠ” мЎ°м§Ғ(functioning tissue)мқҳ лӘЁлҚёлЎң нҶөн•© мӢңнӮӨлҠ”к°Җ мқҙлӢӨ. м„ёнҸ¬лҠ” мЎ°м§Ғмқҳ л№Ңл”© лё”лЎқ(building block)мқҙл©°, мҡ°лҰ¬к°Җ л…јмқҳн•ң мғҲлЎңмҡҙ кё°мҲ мқҖ 3D, мқём ‘н•ң м§Җм—ӯм—җм„ңмқҳ л§өн•‘, лҳҗлҠ” кі„нҶө нҠёлҰ¬(lineage tree)мқҳ 추м Ғмқ„ к°ҖлҠҘн•ҳкІҢ н• мҲҳ мһҲлӢӨ. лҢҖмӮ¬ л¬јм§Ҳ, мӢ нҳё 분мһҗ л°Ҹ м„ёнҸ¬ л°– л§ӨнҠёлҰӯмҠӨ(extracellular matrix)мқҳ кө¬м„ұ мҡ”мҶҢмҷҖ к°ҷмқҖ н•өмӢ¬м Ғмқё м„ёнҸ¬ к°„ 분мһҗ л§Ҳм»ӨлҘј мёЎм •н•ҳкі лӘЁлҚёл§Ғн•ҳлҠ” л°©лІ•мқҖ, лӢЁмқј м„ёнҸ¬лҘј мЎ°м§Ғмқҳ кІ°н•© лӘЁлҚёлЎң лӘЁмңјкё° мң„н•ҙ мӨ‘мҡ”н•ҳлӢӨ. м„ёнҸ¬ мғҒнғңмқҳ лӘЁлҚёкіј 비мҠ·н•ҳкІҢ, л¬ҳмӮ¬н•ң мЎ°м§Ғ лӘЁлҚёмқҖ мЎ°м§Ғмқҳ кі м°Ёмӣҗ кө¬м„ұкіј кё°лҠҘмқ„ мқҙн•ҙн•ҳкі мҳҲмёЎн•ҳкё° мң„н•ң м¶ңл°ңм җмқј лҝҗмқҙлӢӨ. мғҒнҳё мһ‘мҡ©н•ҳлҠ” м„ёнҸ¬мқҳ кіөлҸҷмІҙм—җ мқҳн•ҙ мҲҳн–үлҗҳлҠ” н”„лЎңм„ёмҠӨмқҳ ліөмһЎм„ұкіј 분мһҗм Ғ кІ°м •(molecular decision)мқҖ нһҳл“Өм–ҙ ліҙмқҙм§Җл§Ң, лҸ…нҠ№н•ң кё°лҠҘмқ„ к°Җ진 'мЎ°м§Ғ лӘЁл“Ҳ'мқҳ 분мһҗ-н•ҙл¶Җн•ҷ мғҒмқҳ л§Өк°ң лӢЁкі„к°Җ мЎҙмһ¬н• к°ҖлҠҘм„ұмқҙ мһҲлӢӨ. к·ёлҹ¬лҜҖлЎң, мҡ°лҰ¬лҠ” м„ёнҸ¬к°„мқҳ кіөк°„ к·јм ‘м„ұ л°Ҹ 분мһҗм Ғ мҶҢнҶө(communication)м—җ лҢҖн•ң м •ліҙлҘј м„ёнҸ¬ мғҒнғңм—җм„ң мғҒнҳё мһ‘мҡ©мқҳ кё°лҠҘм Ғ мҳҒн–Ҙкіј кІ°н•©н•ҳм—¬, мЎ°м§Ғм—җм„ң мһ¬л°ңм„ұ лӢӨмӨ‘ м„ёнҸ¬ лӘЁл“Ҳмқ„ к·ңлӘ…н•ҳкі м—°кө¬н•ҳлҠ” н”„л Ҳмһ„мӣҢнҒ¬лҘј кө¬мғҒн•ңлӢӨ. мқҙлҹ¬н•ң лӘЁл“ҲмқҖ мқҙм „м—җ м ңм•Ҳлҗң л°”мҷҖ к°ҷмқҙ, мӨ‘мҡ”н•ң мғҒліҙ кё°лҠҘмқҙ мһҲлҠ” м„ёнҸ¬мҷҖ, мӨ‘мҡ”н•ң м§Җмӣҗ кё°лҠҘ(support function)мқ„ м ңкіөн•ҳлҠ” мЎ°м§ҒмңјлЎң кө¬м„ұ лҗ мҲҳ мһҲлӢӨ(мҳҲ. м§Җл°©мқҳ м§Җл°©(adipocyte) м„ёнҸ¬, к·јмңЎмқҳ к·јм„ёнҸ¬(myocyte) лҳҗлҠ” мһҘмқҳ м җл§үмқҳ мғҒн”ј м„ёнҸ¬(epithelial cell)). мқҙлҹ¬н•ң лӘЁл“Ҳмқҙ мӢӨн—ҳм ҒмңјлЎң нҠ№м„ұнҷ”лҗҳкі м—°кө¬лҗ мҲҳ мһҲмқ„ л•Ң, лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷмқҖ мғқл¬јн•ҷмқҳ к·јліём Ғмқё мқҙн•ҙм—җм„ң мӢӨм ң нҳҒлӘ…мңјлЎң мқҙлҒҢ мҲҳ мһҲлӢӨ.
...................(кі„мҶҚ)
м¶ңмІҳ : мғқл¬јн•ҷм—°кө¬м •ліҙм„јн„°(BRIC) (л°”лЎңк°Җкё°)
|
IP : 192.168.31.*** 
] нҳ„мғҒн•ҷл¶Җн„° л©”м»ӨлӢҲмҰҳк№Ңм§Җ лӢЁмқј м„ёнҸ¬ мң м „мІҙн•ҷмқҳ нҷ•мһҘ кІҢмӢңл¬јлЎң л°”лЎңк°Җкё°,QRмҪ”л“ң")
|
| лІҲнҳё | нҢҢмқј | м ңлӘ© | мқҙлҰ„ | мһ‘м„ұмқј | мЎ°нҡҢмҲҳ |
|---|---|---|---|---|---|
| 455 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2020.01.02 | 12,456 | ||
| 454 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.10.14 | 27,594 | ||
| 453 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.08.28 | 13,623 | ||
| 452 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.08.28 | 11,776 | ||
| 451 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.08.08 | 13,080 | ||
| 450 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.08.08 | 13,650 | ||
| 449 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.08.08 | 12,528 | ||
| 448 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.07.25 | 13,036 | ||
| 447 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.07.25 | 13,269 | ||
| 446 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.07.25 | 11,949 | ||
| 445 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.06.03 | 16,051 | ||
| 444 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.05.29 | 17,073 | ||
| 443 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.05.22 | 16,537 | ||
| 442 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.05.22 | 16,232 | ||
| 441 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.04.01 | 16,687 | ||
| 440 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2019.04.01 | 17,687 | ||
| 439 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2018.05.23 | 29,638 | ||
| 438 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2018.05.23 | 18,230 | ||
| 437 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2018.05.21 | 21,575 | ||
| 436 | кё°мҲ мӮ¬м—…нҷ”нҢҖ | 2018.05.09 | 17,149 | ||